
兰索拉唑肠溶胶囊溶出一致性研究
2021-01-08 01:33李浩然张玉倩张志清
食品与药品 2020年6期
李浩然,张玉倩,康 宇,曹 培,张志清
(河北医科大学第二医院药学部,河北 石家庄 050000)
兰索拉唑[1-2]是继奥美拉唑之后新一代的质子泵抑制剂,其生物利用度和抑制胃酸分泌作用相较于奥美拉唑有明显提高,主要用于胃溃疡、十二指肠溃疡、吻合口溃疡、卓-艾综合征和反流性食管炎等疾病治疗[3-6]。兰索拉唑肠溶胶囊口服易吸收,患者顺应性好,广泛用于治疗胃溃疡等相关疾病。目前,我国除了日本武田的原研药外,还有许多兰索拉唑肠溶胶囊仿制药,且国产的仿制药尚未进行仿制药的质量与疗效一致性评价。
溶出行为是口服固体制剂质量的关键指标之一,口服固体制剂的吸收取决于药物在胃肠道中的释放和溶解,药物的溶出对其在体内的吸收有重要影响,可通过体外溶出试验预测其体内行为[7]。因此,本实验考察了7个兰索拉唑肠溶胶囊仿制药在4种不同介质中的溶出行为,并绘制溶出曲线,以f2相似因子法评价溶出曲线的相似性,旨在对兰索拉唑肠溶胶囊国产仿制药的体外溶出进行一致性评价。
1 仪器与材料
1.1 仪器
Waterse2695高效液相色谱仪(美国Waters);2489 UV/Vis检测器(美国Waters);CPA225D型电子分析天平(德国Sartorius);ZRS-4型智能溶出试验仪(天津大学无线电厂);pH 213型台式酸度计(罗马尼亚HANNA)。
1.2 药品与试剂
兰索拉唑对照品(中国食品药品检定研究院,批号:100709-201705);兰索拉唑肠溶胶囊[原研药:天津武田(A厂家),批号0243A;仿制药: B厂家,批号190202;C厂家,批号18120203;D厂家,批号190105; E厂家,批号AHKG04;F厂家,批号1190702;G厂家,批号20190312; H厂家,批号190702]。甲醇(赛默飞世尔,分析纯,批号:195211);浓盐酸(康德化工,批号:20181106);磷酸二氢钾(天津永大,批号:YD20190712);氢氧化钠(北京益利,批号:20180217)。
2 方法与结果
2.1 色谱条件
色谱柱:Diamonsil C18(250 mm×4.6 mm,5 μm);流动相:甲醇-水(73:27);流速:1 ml/min;柱温:30 ℃;检测波长:285 nm;进样量:20 μl。
2.2 方法学考察
2.2.1 专属性试验 按2.1项下色谱条件,分别取流动相溶液、兰索拉唑对照品溶液及供试品溶液20 μl测定,记录色谱图。本实验条件下,色谱峰分离良好,辅料对兰索拉唑的测定无干扰,该方法专属性良好,结果见图1。

图1 HPLC图谱
2.2.2 线性试验 取兰索拉唑对照品约10 mg,精密称定,置入100 ml量瓶,用含0.01 mol/ml氢氧化钠的流动相溶液稀释至刻度,得104.2 μg/ml对照品溶液,逐级稀释,配制浓度分别为1.042,2.084,4.168,8.336,16.672,33.334 μg/ml系列对照品溶液,按2.1项下色谱条件测定,记录峰面积。以峰面积Y为纵坐标,浓度X(μg/ml)为横坐标,进行线性回归,得回归方程Y=51 370X-5640.3(r2=0.999),兰索拉唑在1.042~33.334 μg/ml浓度范围内线性关系良好。
2.2.3 加样回收率试验 取含量已知的兰索拉唑肠溶胶囊10粒,去除胶囊外壳后精密称定内容物,将内容物研磨成细粉,分别取适量细粉9份(约含对照品10 mg)精密称定,置入50 ml量瓶,流动相溶解并稀释至刻度,摇匀滤过。分别精密量取每份滤液4 ml,置入50 ml量瓶,然后精密加入200.6 μg/ml对照品溶液2,3.2,4 ml,流动相稀释至刻度,每个浓度制备3份,按2.1项下色谱条件测定,以加入量和测得量计算回收率,结果为100.17 %,100.32 %,99.93 %,RSD为0.40 %,0.37 %,0.67 %,加样回收率良好。
2.2.4 精密度试验 配制浓度为8.336 μg/ml对照品溶液,同日及连续3天分别连续测定5份该对照品溶液,记录峰面积,并计算日内精密度RSD为0.24 %,日间精密度RSD为0.61%,表明该方法精密度良好。
2.2.5 稳定性试验 兰索拉唑是苯并咪唑类化合物,显碱性,在酸性条件下易分解[8-9],因此在溶出试验中从不同介质取样后需加入一定量的氢氧化钠增加其稳定性。以pH 4.5,pH 6.0和pH 6.8的磷酸盐缓冲液分别配制浓度为8.336 μg/ml的对照品溶液,精密量取2 ml并加入0.1 mol/L氢氧化钠溶液1 ml(n=6),放置0,1,2,3,5,7,9 h后按2.1项下色谱条件测定,计算得RSD为0.59 %,0.69 %,0.38 %,表明兰索拉唑在加入氢氧化钠溶液后的pH 4.5,pH 6.0和pH 6.8磷酸盐缓冲液中放置9 h稳定性良好。
2.3 溶出度考察
2.3.1 不同介质中的稳定性考察 分别以pH 4.5,pH 6.0,pH 6.8磷酸盐缓冲液为溶媒制备一定浓度的对照品溶液,将制备好的对照品溶液放置0,15,30,45,60 min后,按2.1项下色谱条件测定,考察其在不同介质中的稳定性,结果表明,兰索拉唑在3种介质中的稳定性符合测定要求,因此pH 4.5,pH 6.8和pH 8.0的磷酸盐缓冲液可作为兰索拉唑肠溶胶囊的溶出介质,见表1。
2.3.2 溶出介质的制备 按表2量取试剂置入1000 ml量瓶,蒸馏水稀释至刻度,摇匀即得。
2.3.3 pH 1.2盐酸溶液中溶出度考察 取兰索拉唑肠溶胶囊放入转篮,按溶出度与释放度测定法(通则0931第一法 方法2)测定[10](n=12),以37 ℃1000 ml pH 1.2盐酸溶液为溶出介质,转速为100 r/min,120 min时,取出转篮,按2.1项下方法测定篮内兰索拉唑的剩余量,间接计算兰索拉唑肠溶片在pH 1.2盐酸溶液中的溶出度,绘制溶出曲线,结果表明,兰索拉唑肠溶胶囊在pH 1.2盐酸溶液中无溶出行为,见图2。
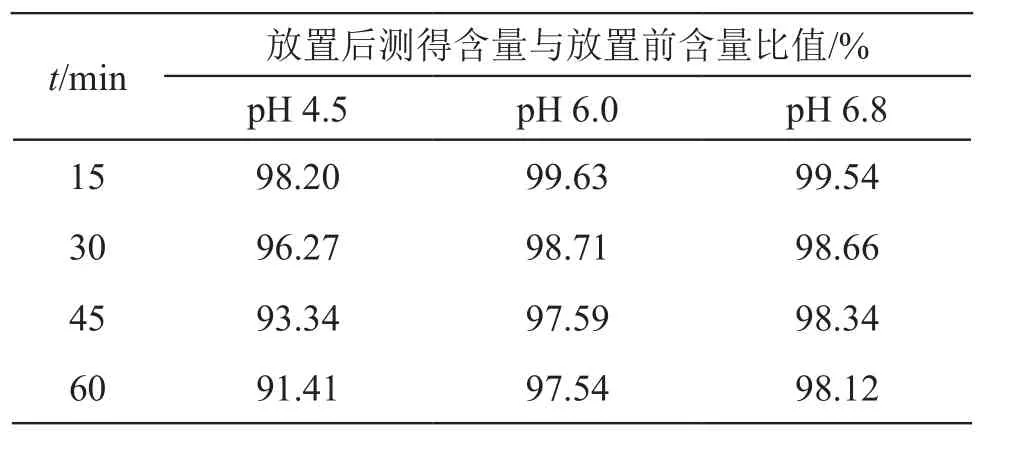
表1 兰索拉唑在不同pH溶出介质中的稳定性
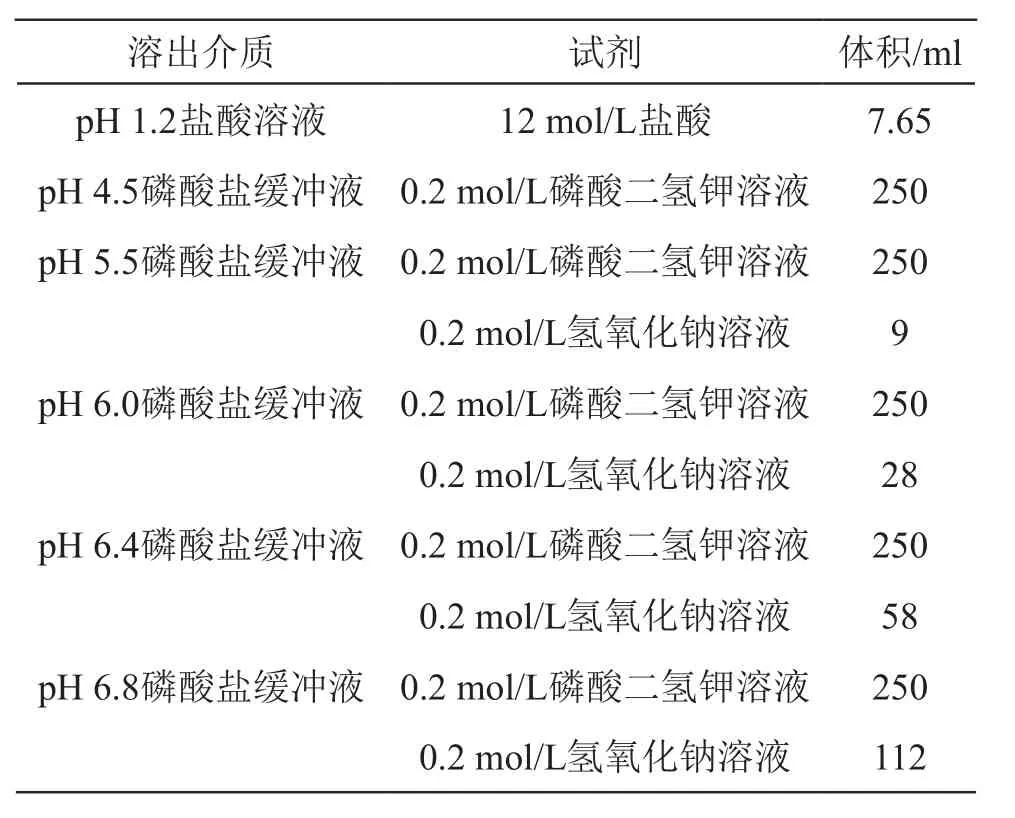
表2 不同pH溶出介质的制备
2.3.4 磷酸盐缓冲液中溶出度考察 取兰索拉唑肠溶胶囊放入转篮(n=12),以37 ℃ 1000 ml pH 1.2盐酸溶液为溶出介质,转速为100 r/min,120 min时,将盐酸溶液弃去,加入预热至37 ℃的磷酸盐缓冲液1000 ml(pH 4.5或pH 5.5或pH 6.0或 pH 6.4或pH 6.8),转速不变,分别于5,10,15,20,30,45 min取溶液3 ml滤过,同时补充相同pH、温度和体积的磷酸盐缓冲液,精密量取续滤液2 ml,立即加入0.1 mol/L氢氧化钠溶液1 ml,摇匀,按2.1项下色谱条件测定,记录峰面积,按标准曲线计算药物浓度,计算药物溶出度并绘制药物溶出曲线,结果见图2。由结果可见,兰索拉唑肠溶胶囊原研药与仿制药在pH 6.8磷酸盐缓冲液中有较快的溶出行为,15 min的溶出量达85 %以上;而在pH 5.5,pH 6.0和pH 6.4磷酸盐缓冲液中仿制药的溶出行为偏慢,且与原研药有不同程度差异,在pH 4.5磷酸盐缓冲液中仿制药无溶出行为,原研药仅有少量的药物溶出。
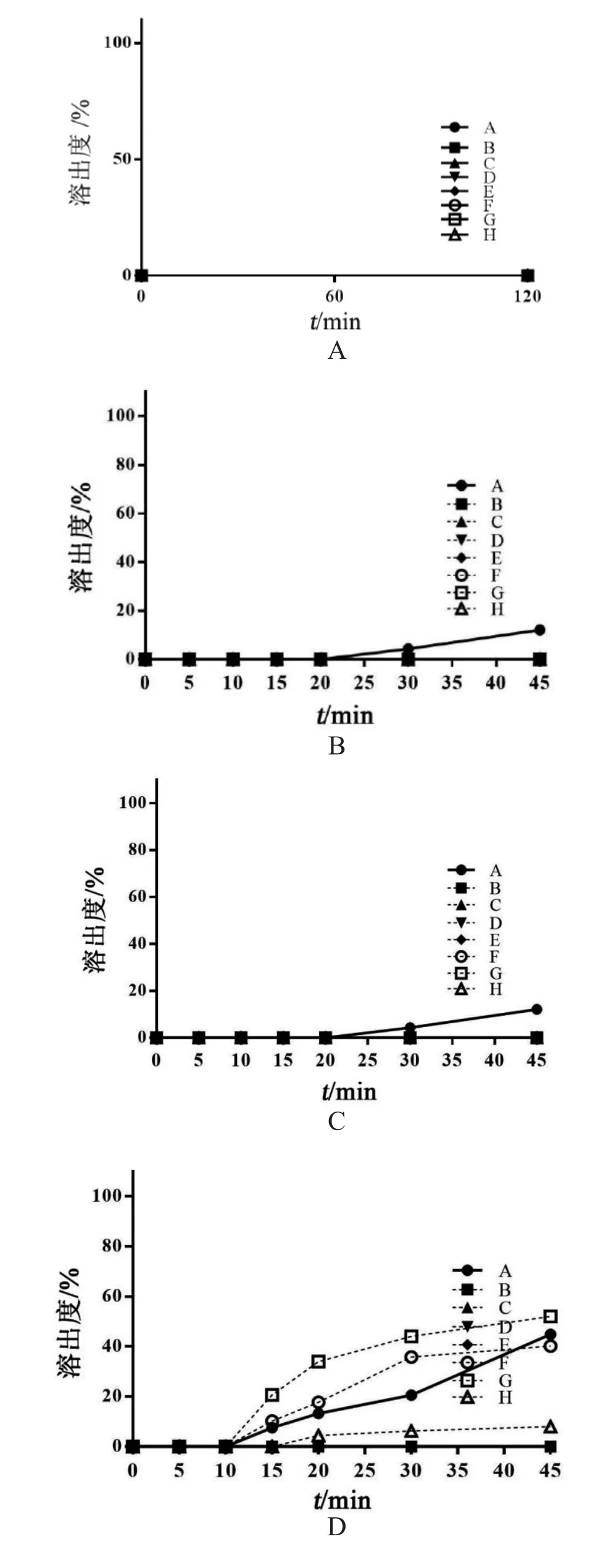
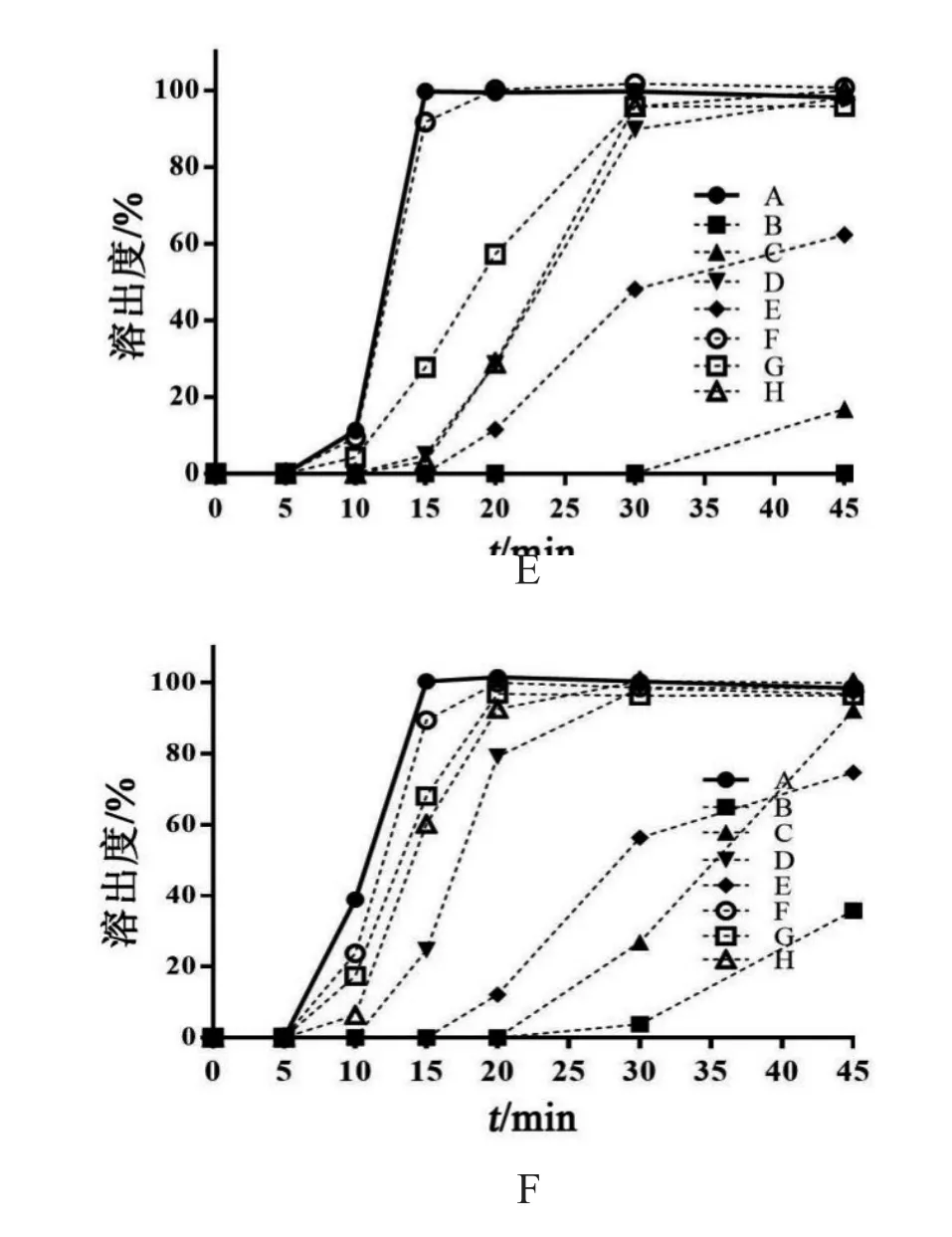
图2 兰索拉唑肠溶胶囊在不同介质中的溶出曲线
2.3.5 溶出曲线相似性比较 采用f2相似因子法比较溶出曲线的相似性[11],当f2≥50时,认为两条溶出曲线具有相似性,结果见表3。另外,当原研药和仿制药在15 min内的药物溶出量≥85 %时,可认为两者溶出行为相似,无需进行f2比较。本实验中,兰索拉唑肠溶胶囊原研药和仿制药在pH 6.8磷酸盐缓冲液中15 min时的药物溶出量在85 %以上,因此认为该仿制药与原研药在pH 6.8磷酸盐缓冲液中溶出行为一致。由结果可见,兰索拉唑肠溶胶囊仿制药在pH 1.2盐酸溶液和pH 4.5磷酸盐缓冲液中均无药物释放,与原研药的溶出行为一致。在pH 5.5,pH 6.0和pH 6.4磷酸盐缓冲液中仅有F厂家仿制药的溶出曲线与原研药相似,其余仿制药与原研药的溶出曲线均有较大差异。因此,在7个仿制药中仅有F厂家的溶出行为与原研药一致。
3 讨论
pH 6.0磷酸盐缓冲液能较好地区分不同厂家兰索拉唑肠溶胶囊的溶出行为,仿制药在pH 6.0磷酸盐缓冲液的溶出量比原研药少,溶出速率慢,可能与该制剂的辅料和制备工艺有关。原研药在pH 6.0、pH 6.4和pH 6.8介质中均能较快释药,而仿制药在两种介质中的释药行为有较大差异,表明该仿制药肠溶衣的临界溶点可能接近或高于pH 6.0[12],因此在pH 6.0介质中该肠溶衣溶解慢或不溶解导致释药量减少。

表3 溶出曲线相似性比较结果
机体正常肠道环境一般为pH 6.8,但在胃酸分泌过多等不同疾病状态下可能使部分肠道环境的pH降低[13]。因此,本实验以pH 5.5,pH 6.0,pH 6.4磷酸盐缓冲液模拟不同人群的肠道环境。在不同介质中兰索拉唑肠溶胶囊仿制药的释药量和释药速率与原研药有不同程度的差异,表明该药在同样条件肠道中的释药行为也可能不同,可能是导致该仿制药治疗效果与原研药存在差异的原因之一。因此,建议兰索拉唑肠溶胶囊仿制药厂家重视体外溶出行为在一致性研究中的重要作用,深入研究制剂工艺辅料应用,早日达到一致性评价的制剂标准。
猜你喜欢
中国医院用药评价与分析(2022年5期)2022-06-23
家庭医药·快乐养生(2022年6期)2022-06-23
中国典型病例大全(2022年13期)2022-05-10
医学概论(2022年4期)2022-04-24
空间科学学报(2021年1期)2021-05-22
大众健康(2019年6期)2019-06-10
疯狂英语·新读写(2018年3期)2018-11-29
西南军医(2016年5期)2016-01-23
中国药业(2014年17期)2014-05-26
