
硼元素含量对非晶态合金Co-Ni-B磁性的影响
2020-12-28 02:24方志刚秦渝张伟廖薇许友
河北大学学报(自然科学版) 2020年6期
方志刚,秦渝,张伟,廖薇,许友
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
一直以来,Co系与Ni系非晶态合金因具有优异的物理化学性能以及在多方面的应用前景而成为当下最受欢迎的研究热点之一[1-4].近年来,研究者们在过渡金属Co、Ni的催化性能方面有了不错的进展[5-6],在磁学性能方面更是取得了巨大的成就[7-8].例如Wu等[9]采用TR-MOKE技术研究了垂直Co/Ni基SAF结构中的磁动力学,从而探究层间耦合对[Ni/Co]4/Ru(tRu)/[Co/Ni]3垂直磁性薄膜磁化动力学的影响,研究结果为自旋电子学应用中的垂直交换耦合系统的磁动力学提供了新的见解;Barla等[10]观察到外延石墨烯层介导的单个Co原子与Ni之间吸附位置依赖性主要是AFM交换耦合,且Co原子具有很强的平面外磁各向异性,该研究结果对于利用石墨烯作为磁性异质结构和自旋电子器件的间隔层以及理解石墨烯与磁性杂质之间的复杂相互作用具有重要意义.
而具有开创性的Stern-Gerlach实验作为量子理论和原子物理学中的一个重要里程碑,它涉及到自旋粒子与外加磁场梯度之间的相互作用.现如今,许多科研人员从不同角度对Stern-Gerlach实验进行研究以期进一步完善该实验的相关内容.Rohrmann等[11]利用Stern-Gerlach实验研究了Fe@Sn12团簇的磁效应,从束流剖面的位移中提取出磁偶极矩的大小,从而为电子轨道角动量对磁偶极矩的贡献(部分猝灭)提供了证据;Håkan等[12]则表示只有将产生磁场梯度的磁性材料的波动磁场所产生的横向自旋弛豫考虑在内,才能充分理解Stern-Gerlach实验.
尽管过渡金属Co、Ni元素优异的磁学性能正逐渐被人们发掘,但仍有许多关键且重要的问题尚待人们去挖掘和探究.截至目前,从电子自旋态密度角度出发对团簇进行深入研究以获得不同轨道电子自旋的具体分布情况还鲜少有人探究.因此,本文在已有Co-Ni原子比例模型中掺杂不同含量的类金属B,以分别构建出单硼模型Co3NiB与双硼模型Co3NiB2,并从其s、p、d轨道上未成对电子数和态密度入手进行研究,以完善Stern-Gerlach实验的相关内容,同时也为今后研究三元非晶态合金Co-Ni-B体系提供有价值的理论依据.
1 模型建立与理论计算方法
1.1 模型的选择与建立
为研究非晶态合金Co-Ni-B体系的磁学性质,本文基于已有的Co-Ni原子比例[13],选择不同B原子数目以探究掺杂不同B元素含量对Co-Ni-B体系磁性的影响.为此,本文选择奇数为1、偶数为2的B原子数为代表,分别设计出单硼团簇模型Co3NiB与双硼团簇模型Co3NiB2,通过微观角度研究这2个团簇的磁学性能,以期为研究非晶态合金Co-Ni-B体系的相关性质提供一定的理论参考.
1.2 理论计算方法
基于较高量子化学计算水平B3LYP/Lanl2dz下,依据拓扑学原理[14]运用密度泛函理论[15]分别对单硼模型Co3NiB和双硼模型Co3NiB2的所有初始构型在不同自旋多重度情况下进行全参数优化和相关频率计算,对计算所得优化构型的非金属B原子采用Dunning/Huzinaga双ξ基组(9s,5p/3s,2p),对过渡金属Co、Ni原子采用18-eECP的双ξ基组(3s,3p,3d/2s,2p,2d)[16].文中所有的计算过程均运用Gaussian09程序在启天M7150计算机上完成,其中与磁性相关的计算全部使用Multiwfn程序[17]完成.
2 实验结果与数据分析
2.1 团簇Co3NiB与Co3NiB2的优化构型
对将团簇Co3NiB与Co3NiB2各异构体中含虚频的不稳定构型和相同构型排除之后的剩余稳定构型进行异构化转化,最终共得到如图1所示的8种优化构型.其中,团簇Co3NiB与Co3NiB2各4种.团簇Co3NiB为单、三重态,其优化后的稳定构型主要以中心平面四边形(1(3))和三角双锥(2(3)、3(3)与1(1))两类构型存在.团簇Co3NiB2为二、四重态,其优化后的稳定构型主要以五棱锥(1(4)与1(2))、单帽四棱锥(2(4))及单帽三角双锥(2(2))三类构型存在.
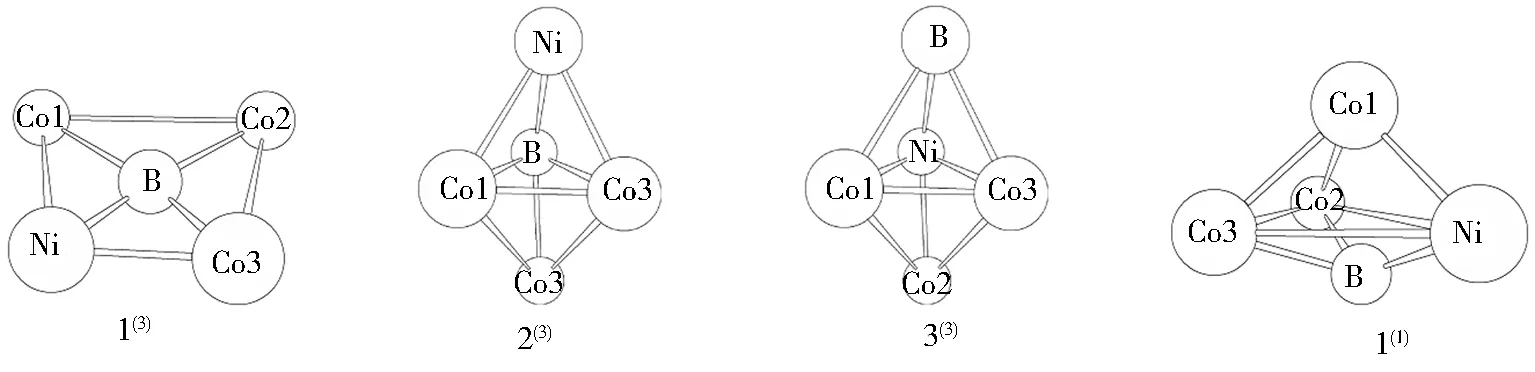
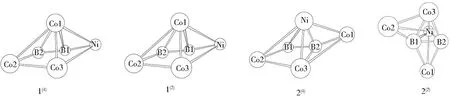
图1 团簇Co3NiB与Co3NiB2的优化构型Fig.1 Optimized configurations of cluster Co3NiB and Co3NiB2
2.2 单硼团簇模型Co3NiB的磁学性质
团簇不同原子轨道中未成对电子的数目及其自旋运动情况是影响非晶态合金体系磁学性质的重要因素.因此,对非晶态合金Co-Ni-B体系不同原子的成单电子数和电子自旋运动情况进行深入探究是十分必要的.根据自旋多重度的定义,单重态构型原子核外无成单电子,故其不能展现出团簇的磁学性质.为此,本文通过研究团簇Co3NiB含有2个成单电子的三重态构型和团簇Co3NiB2分别含有1个及3个成单电子的二、四重态构型来探究不同B元素含量对非晶态合金Co-Ni-B体系磁学性质的影响.
表1为团簇Co3NiB s、p、d轨道上的成单电子数.其中,正值表示净剩电子为自旋向上的α电子,负值表示净剩电子为自旋向下的β电子.不难发现,单硼团簇模型d轨道上的净剩电子均为自旋向上的α电子,且其d轨道上的未成对电子数明显高于s、p轨道,说明在团簇Co3NiB中,磁性主要由d轨道贡献.
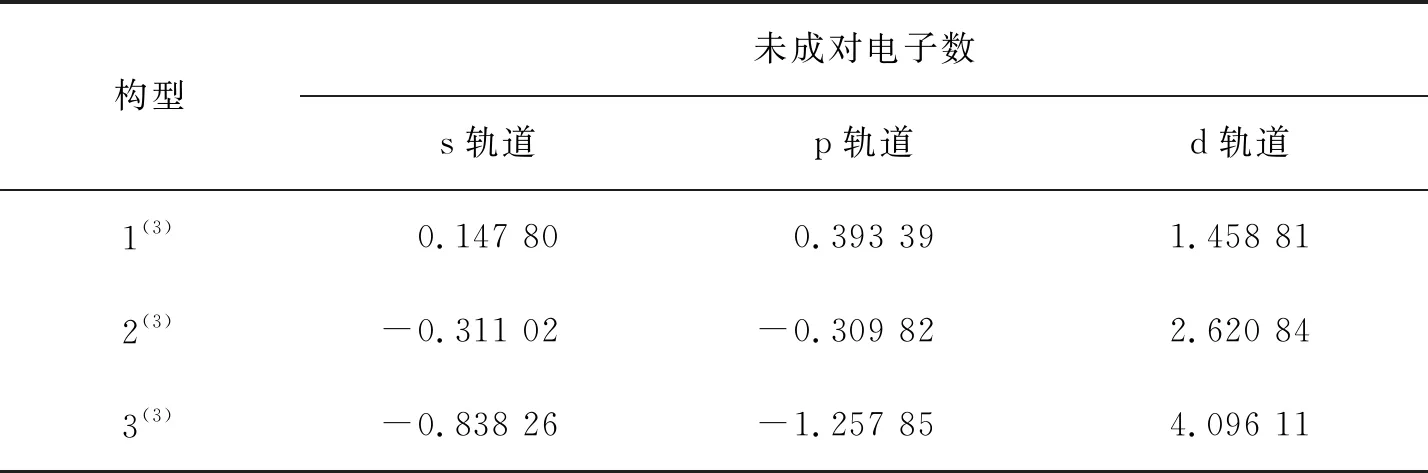
表1 团簇Co3NiB的s、p、d轨道未成对电子数
从另一个角度来看,团簇Co3NiB的磁性主要由自旋向上的α电子贡献,那么自旋向下的β电子对于团簇磁性而言是具有一定的削弱作用.仔细比较图2中各优化构型s、p轨道上的成单电子数发现,构型1(3)p轨道上α电子数目明显多于s轨道,说明在构型1(3)中,p轨道对团簇磁性的贡献大于s轨道.同为三角双锥构型的2(3)与3(3)的s、p轨道上均为自旋向下的β电子,其中较为稳定的构型2(3)s轨道上的β电子数多于p轨道,说明构型2(3)p轨道对团簇磁性的贡献比s轨道更大,而稳定性较差的构型3(3)则与之完全相反,其s轨道对团簇磁性的贡献比p轨道大.综上分析可知,在单硼团簇模型中,d轨道是团簇磁性的主要贡献者,p轨道次之,而s轨道对团簇磁性的贡献最小.即各轨道对单硼团簇模型Co3NiB优化构型磁性的贡献由大到小依次为d轨道>p轨道>s轨道.
为更直观地展示团簇不同原子对非晶态合金体系磁性的贡献,图3给出了团簇Co3NiB各优化构型不同原子磁矩的具体分布情况.由图3可知,团簇磁性主要由d轨道上自旋向上的α成单电子贡献,表明其磁矩为正.结合图3可发现团簇Co3NiB的整体磁矩为正,这就意味着如果某原子的磁矩为负值,则该原子对团簇磁性将产生一定的削弱作用.由图3可知,所有优化构型中各原子的磁矩总和均为正值,其中Co原子的磁矩最大且远大于零,说明在团簇Co3NiB中,Co原子是磁性的主要贡献者.除构型1(3)外,其余构型B原子的磁矩均为负值,说明非金属B原子的掺杂在一定程度上削弱了其所研究对象的磁性,这与徐诗浩等[18]提出的“非金属B原子的掺杂导致团簇磁性下降”结论一致.此外发现,在同一重态下的不同构型中,Ni原子的变化趋势也发生了不规则变化,这说明相同原子在同一重态下的不同构型中,对团簇磁性产生的影响不同.
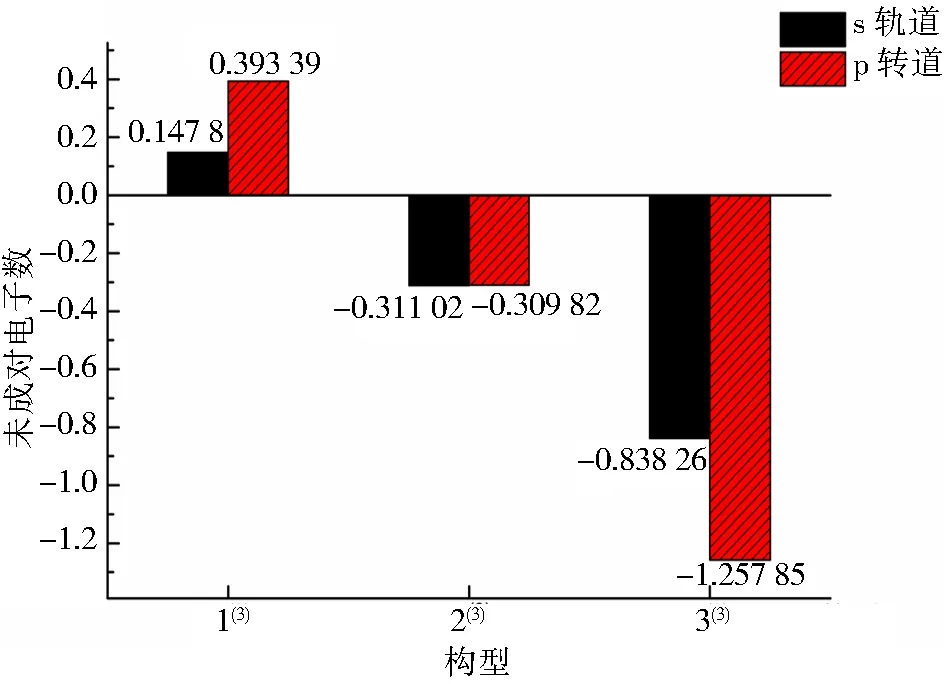
图2 团簇Co3NiB s、p轨道上的未成对电子数Fig.2 Number of unpaired electrons in s, p orbitals of cluster Co3NiB
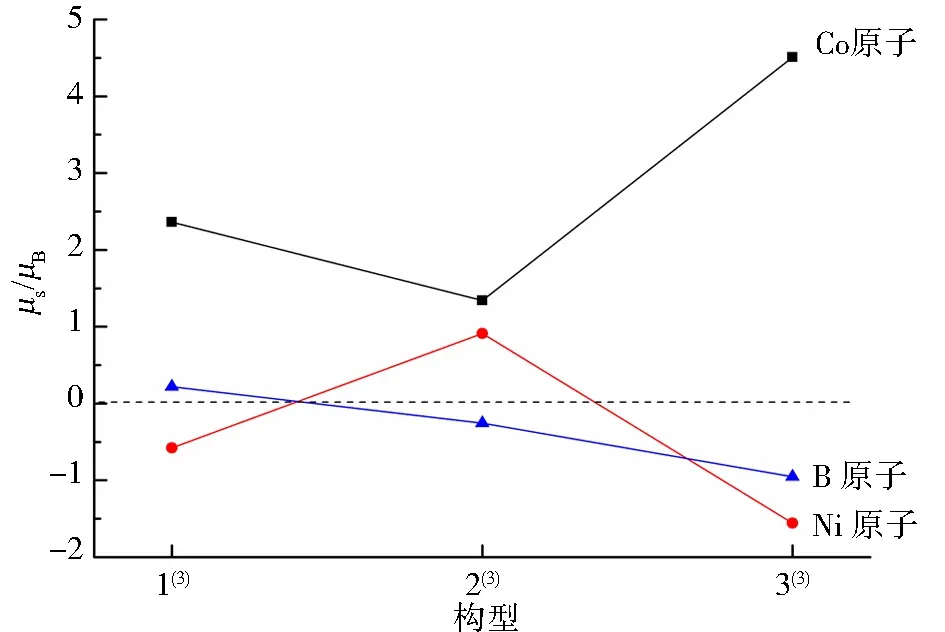
图3 团簇Co3NiB不同原子的磁矩分布Fig.3 Specific distribution of magnetic moments of different atoms in cluster Co3NiB
图4为团簇Co3NiB 3种优化构型s、p、d轨道上电子的自旋态密度图.图中实线表示自旋向上的α电子,虚线表示自旋向下的β电子.α与β电子的曲线对能量的积分和为该轨道的成单电子数,即对应表1中各轨道的未成对电子数.2条曲线的对称性越好,则表明该轨道上的净剩电子数越少,对团簇磁性的贡献就越小.
由图4可知,所有优化构型s、p轨道自旋向上的α电子与自旋向下的β电子均关于横轴呈现出极好的对称性,说明单硼团簇模型Co3NiB的s、p轨道成单电子数均较少,对团簇磁性的贡献较小.不同于s、p轨道,d轨道自旋向上与自旋向下的态密度无论是从形状还是峰值的绝对值大小都展现出了较大的差异,其中自旋向上的α电子对能量的积分明显大于自旋向下的β电子对能量的积分,这不仅进一步证实了“磁性主要由d轨道贡献”的结论,而且还说明了磁性主要是由d轨道上自旋向上的α成单电子贡献.另外,所有优化构型的s、p轨道均有在Z与Z’处发生电子自旋方向改变的情况,其中s轨道发生电子自旋方向改变在0~5 eV内,构型1(3)的p轨道在20~30 eV内发生了电子自旋方向的改变(有且仅有1处峰的电子发生了自旋方向的改变),而构型2(3)与3(3)的p轨道则是在10~20 eV内有2处峰发生了电子自旋方向的改变,这说明针对单硼团簇模型各优化构型内部不同的轨道,使电子自旋方向发生改变的能量值和波峰数目均不一样.
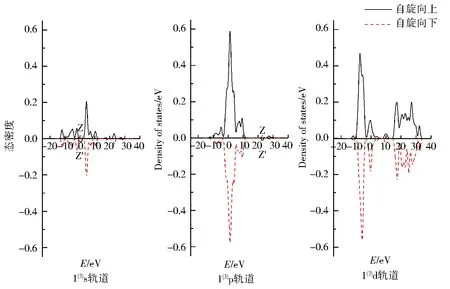
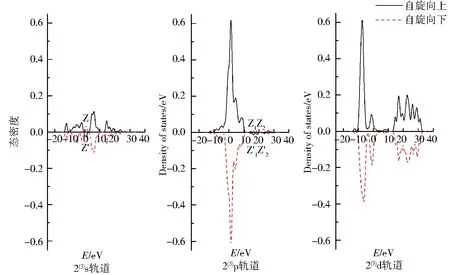
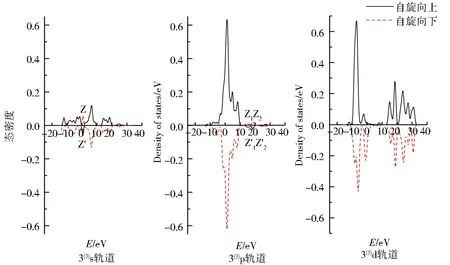
图4 团簇Co3NiB 3种优化构型的s、p、d轨道电子自旋态密度Fig.4 Density of states of electrons spinning in s,p,d orbitals of three optimized configurations of cluster Co3NiB
2.3 双硼团簇模型Co3NiB2的磁学性质
由表2可知,在双硼团簇模型Co3NiB2中,其d轨道上的净剩电子同单硼团簇模型一样均为自旋向上的α电子,且其d轨道上的成单电子数远远高于s、p轨道,说明在团簇Co3NiB2中,磁性同样主要由d轨道贡献.依据图5对团簇Co3NiB2的s、p轨道进行细致分析发现,构型1(4)s轨道上自旋向下的β电子数目明显多于p轨道,说明构型1(4)p轨道对团簇磁性的贡献比s轨道大.对于除构型1(4)外的其余构型而言,它们的s轨道上均为自旋向上的α电子,其中构型1(2)p轨道上的α电子少于s轨道,且构型2(4)与2(2)的p轨道上均为自旋向下的β电子,这说明除构型1(4)外,其余所有构型的s轨道对团簇磁性的贡献均大于p轨道.综上所述,在双硼团簇模型中,d轨道对团簇磁性的贡献最大,s轨道次之,而p轨道贡献最小,即各轨道对双硼团簇模型Co3NiB2优化构型磁性的贡献排布为d轨道>s轨道>p轨道.
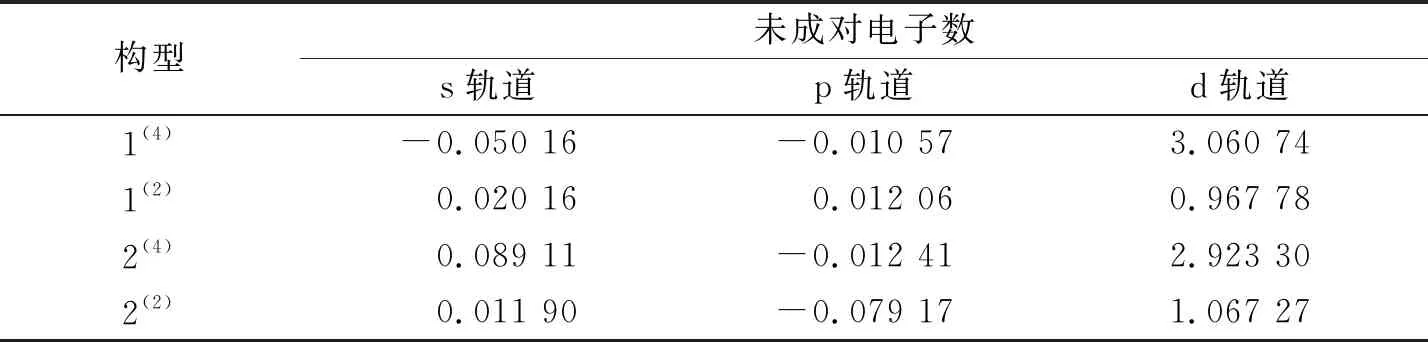
表2 团簇Co3NiB2的s、p、d轨道未成对电子数
由图6可知,团簇Co3NiB2所有优化构型中各原子的磁矩总和同单硼团簇模型Co3NiB一样均为正值,其中Co原子的磁矩最大且远大于零,说明在双硼团簇模型Co3NiB2中,Co原子仍然是磁性的主要贡献者.而团簇Co3NiB2所有优化构型的B原子磁矩均为负值,这进一步说明了非金属B原子的掺杂在一定程度上削弱了其所研究对象的磁性.值得注意的是,在不同重态构型下,Ni原子呈现出锯齿形的变化趋势,其磁矩在四重态时为正值、在二重态时为负值,这说明相同原子在不同重态构型中会对团簇磁性产生不同的影响.
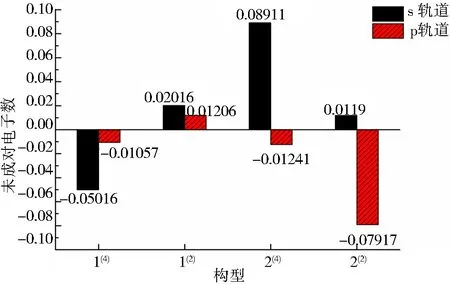
图5 团簇Co3NiB2 s、p轨道上的未成对电子数Fig.5 Number of unpaired electrons in s, p orbitals of cluster Co3NiB2
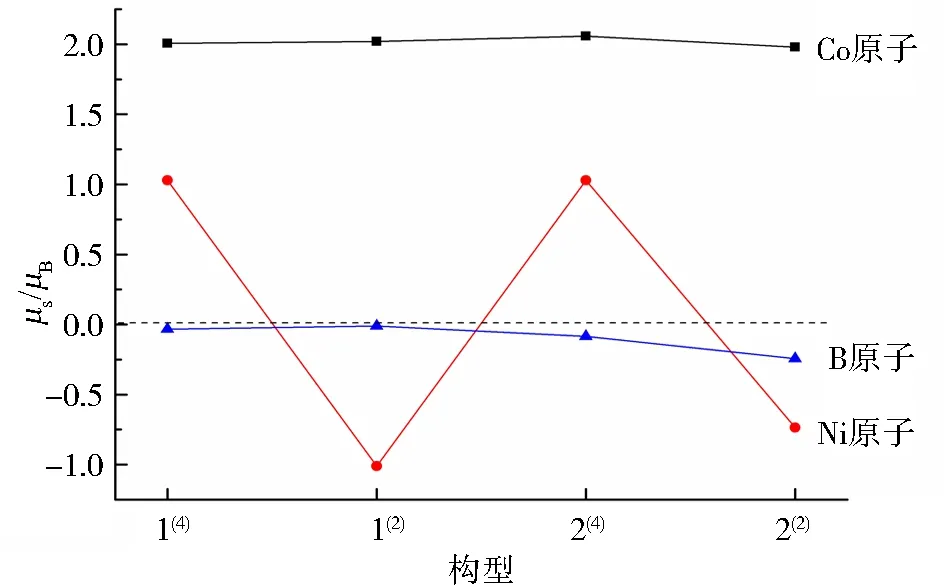
图6 团簇Co3NiB2不同原子的磁矩分布Fig.6 Specific distribution of magnetic moments of different atoms in cluster Co3NiB2
图7为团簇Co3NiB24种优化构型s、p、d轨道上α与β电子的自旋态密度图,其对应的是表2中各轨道的未成对电子数.仔细观察各优化构型的s、p、d轨道发现,所有优化构型s、p轨道与单硼团簇模型一样,其α电子与β电子均关于横轴对称,而d轨道中α电子对能量的积分远远大于β电子对能量的积分,这同样说明了双硼团簇模型的s、p轨道对团簇磁性的贡献较小且磁性主要是由d轨道上自旋向上的α成单电子贡献.与单硼团簇模型相同的是,所有优化构型的s、p轨道均在某处有发生电子自旋方向的改变.不同之处在于双硼团簇模型的s、p轨道都有且仅有1处峰发生了电子自旋方向的改变.其中s轨道发生电子自旋方向改变在0~5 eV内,p轨道则是在20~30 eV内发生了电子自旋方向的改变,这说明针对双硼团簇模型各优化构型内部不同的轨道,使电子自旋方向发生改变的能量值不一样.
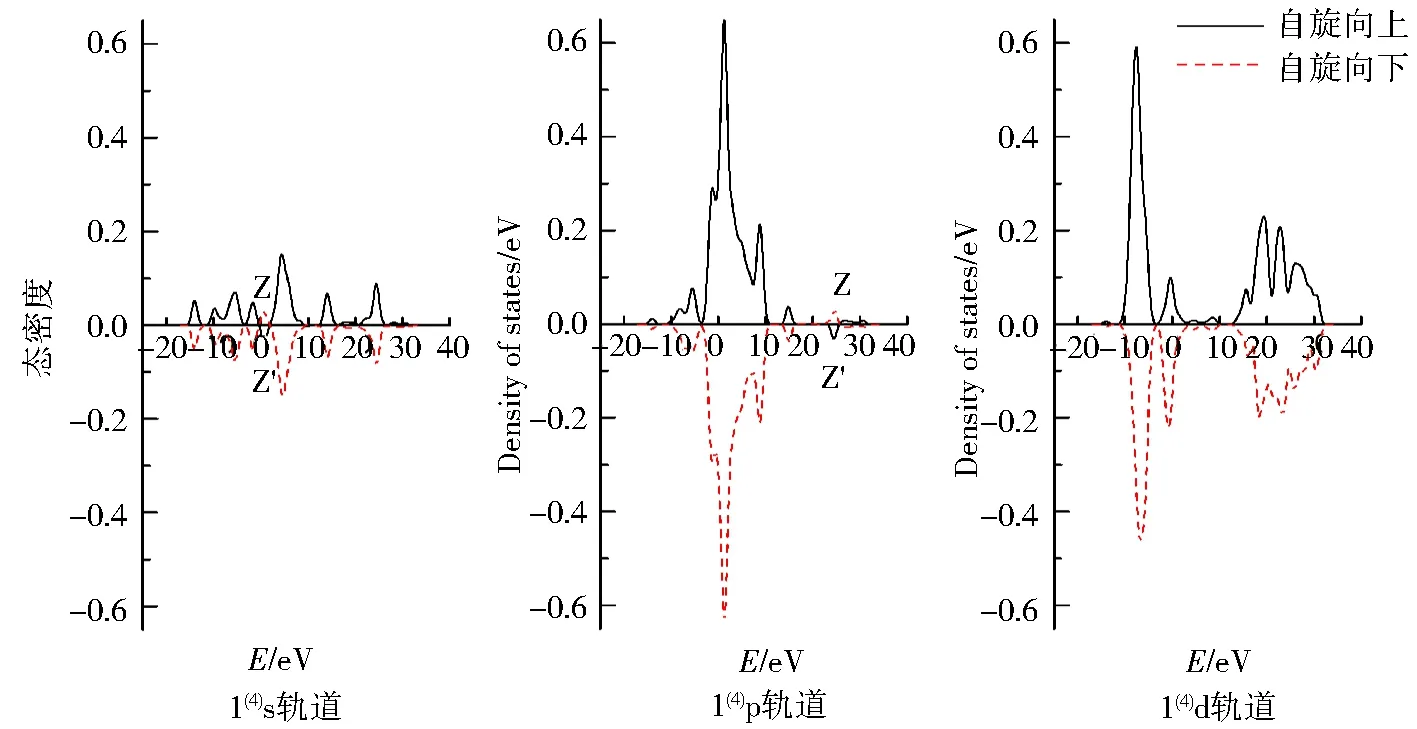
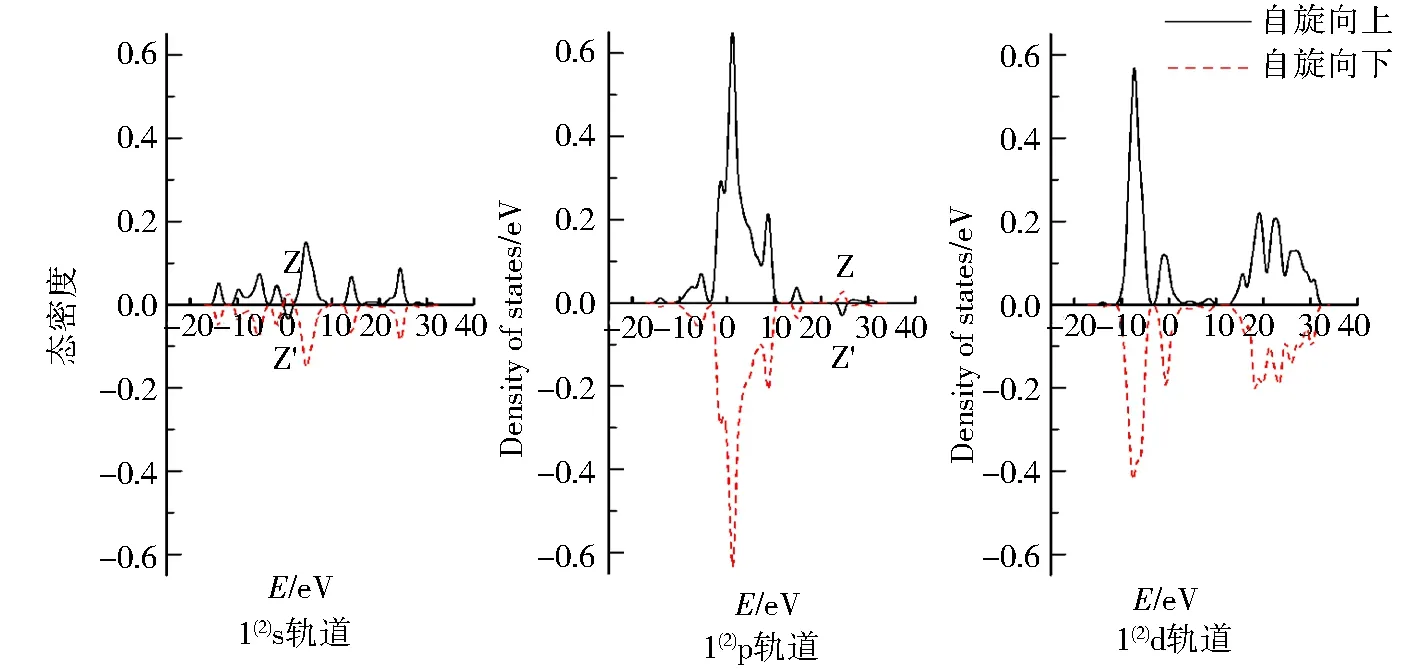
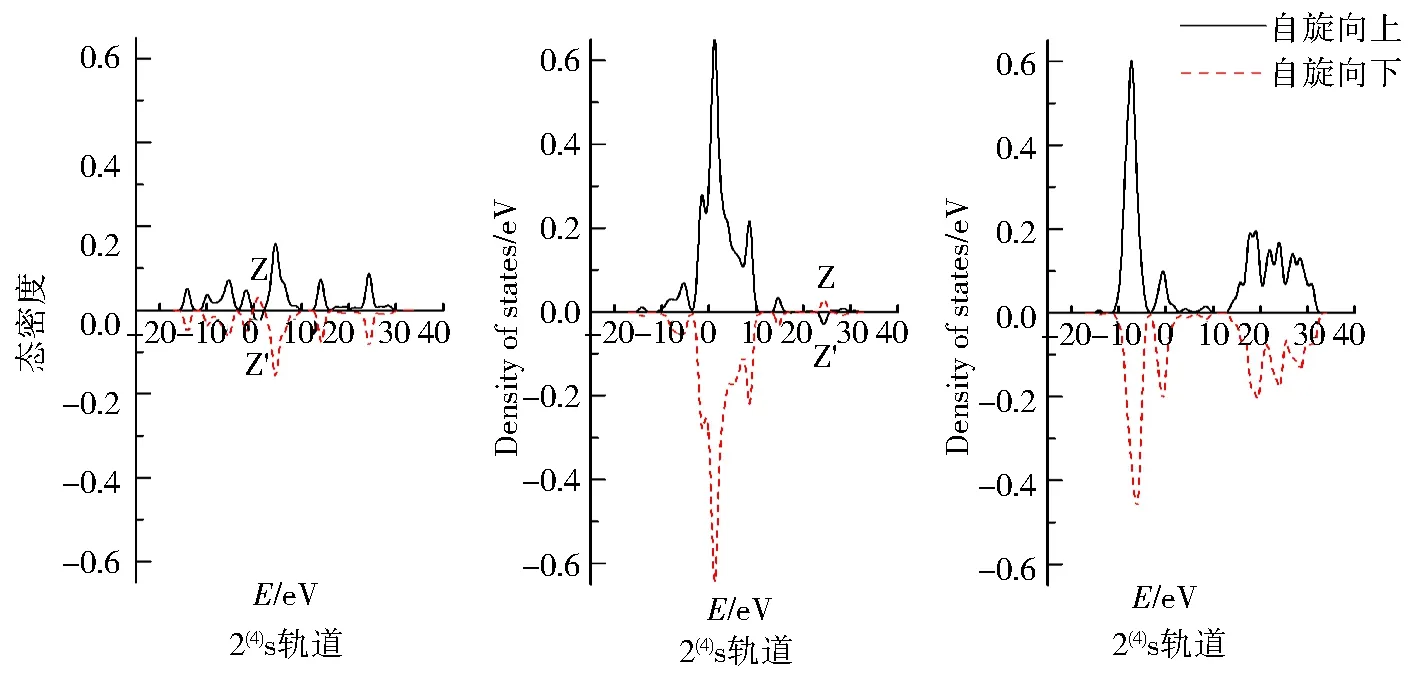
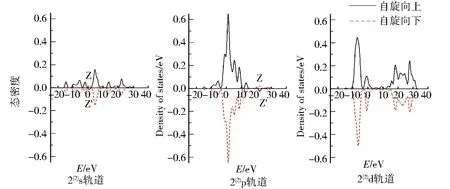
图7 团簇Co3NiB2 4种优化构型的s、p、d轨道电子自旋态密度Fig.7 Density of states of electrons spinning in s,p,d orbitals of four optimized configurations of cluster Co3NiB2
2.4 综合分析
20世纪20年代,德国物理学家奥托·斯特恩和瓦尔特·格拉赫通过Stern-Gerlach实验(Ag原子射线经过非均匀磁场后会分裂成“两条”)首次证实了原子在磁场中取向量子化和原子角动量的量子化.由于作为研究对象的Ag原子最外层电子排布式为4d105s1,因此该实验仅能证明s轨道电子具有自旋向上与自旋向下2个方向,而不能证明p、d轨道上电子的自旋方向是否能发生改变.值得注意的是,在团簇Co3NiB与团簇Co3NiB2所有优化构型s、p轨道的Z与Z′处,α电子与β电子的自旋方向均发生了改变,且针对不同轨道使电子自旋方向发生改变的能量范围也不尽相同.根据量子力学相关理论[19]可知:Stern-Gerlach实验说明了在磁场的立体作用下,电子具有自旋向上和自旋向下的2种能级体现.这不仅验证了原子自旋假设的正确性和原子自旋的空间取向量子化,而且证实了电子具有一种内禀角动量(即自旋角动量),在任意方向的投影只有2个取值的结论.级联Stern-gerlach实验证实了2个力学量不能同时有确定值,即电子自旋的不确定性,这说明自旋向上的α电子与自旋向下的β电子在一定条件下可以相互转化.而团簇Co3NiB与Co3NiB2各优化构型s、p轨道在Z与Z’处发生电子自旋方向改变的现象则很好地为Stern-Gerlach实验与级联Stern-Gerlach实验进行了佐证.
截至目前,在与Stern-Gerlach实验相关的研究成果中尚未发现有人对p、d轨道电子自旋方向是否发生改变进行研究.因此,结合前文分析发现,无论是何种团簇,其s轨道一定会在某处发生电子自旋方向的改变,d轨道则与之相反即一定不会发生电子自旋方向的改变,而从未被探究过的p轨道同s轨道一样,其轨道上电子的自旋方向一定会发生改变(且其所有优化构型的p轨道均能在某处或某2处发生电子自旋方向的改变).同时根据2.2与2.3的分析可知,通过仔细研究各轨道电子自旋态密度图不仅可以发现各轨道电子自旋方向的改变情况,而且还能清晰直观地了解到各轨道各处电子自旋的具体分布情况,这不仅为Stern-Gerlach实验作了十分重要的补充,而且还为完善该实验的研究方法开辟了思路.
3 结论
本文在已有Co-Ni原子比例的基础上,选择不同B原子数目以构建出团簇Co3NiB与Co3NiB2模型,通过对其微观结构的研究来探究出不同B元素含量对非晶态合金Co-Ni-B体系磁学性质的影响.在对团簇Co3NiB与Co3NiB2的磁学性质进行深入探究后,得到如下结论:1)无论是单硼团簇模型还是双硼团簇模型,他们的s、p轨道成单电子数均较少,对团簇磁性的贡献较小;相反,其d轨道上的未成对电子数明显高于s、p轨道且其自旋向上电子的态密度明显大于自旋向下电子的态密度(d轨道自旋向上与自旋向下的曲线不仅均不对称,而且自旋向上曲线对能量的积分明显大于自旋向下曲线对能量的积分),说明在非晶态合金Co-Ni-B体系中,磁性主要由d轨道中的成单电子即自旋向上的α电子贡献,且B元素含量不会改变Co-Ni-B体系磁性的主要贡献轨道和成单电子种类.2)所有优化构型中各原子的磁矩总和均为正值,且不论是单硼团簇模型还是双硼团簇模型.Co原子磁矩均为正值且最大,说明Co原子是非晶态合金Co-Ni-B体系磁性的主要贡献者;除构型1(3)外,所有构型B原子的磁矩均为负值,说明非金属B原子的掺杂在一定程度上削弱了其所研究对象的磁性;而Ni原子磁矩的变化趋势则说明了相同原子在不同构型中对团簇磁性产生的影响不同.3)仔细研究2团簇s、p、d轨道电子自旋态密度图发现,所有优化构型s、p轨道在Z与Z’处的α电子与β电子的自旋方向均发生了改变,且针对不同轨道使电子自旋方向发生改变的能量范围也不尽相同,d轨道一定不会发生电子自旋方向的改变,这从理论上为Stern-Gerlach实验的结论进行了很好的补充.
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
发明与创新·小学生(2019年11期)2019-08-11
电子制作(2018年11期)2018-08-04
军事文摘·科学少年(2017年4期)2017-06-20
北京航空航天大学学报(2017年10期)2017-04-20
中学科技(2015年6期)2015-08-08
