
替代对照品法测定复方甘草口服溶液中甘草苷和甘草酸含量
2020-12-13 10:04魏长勇余敏灵
中国药业 2020年23期
曾 芳,魏长勇,余敏灵
(四川省乐山市食品药品检验检测中心,四川 乐山614099)
复方甘草口服溶液是由甘草流浸膏、愈创甘油醚、复方樟脑酊等制成的复方制剂,镇咳祛痰功效良好。2015年版《中国药典(二部)》中未对甘草苷含量进行控制,本研究中以甘草酸铵为对照品采用高效液相色谱(HPLC)法进行甘草酸含量测定[1]。甘草酸铵易吸潮、价格贵,参考甘草、甘草流浸膏质量标准和相关文献[2-9],以橙皮苷为对照品替代甘草苷、甘草酸铵,采用替代对照品法,以色谱峰保留时间为辅助,并采用紫外光谱特征和纯度检查因子作定性鉴别;在定量分析中,通过测定橙皮苷和被测定物质的相对校正因子,并运用校正因子计算甘草苷和甘草酸在制剂中的含量。现报道如下。
1 仪器与试药
1.1 仪器
1260型高效液相色谱仪(包括DAD检测器,Open-LAB CDS工作站,美国Agilent公司);PE-A10型高效液相色谱仪(包括DAD检测器,珀金埃尔默公司);Ultimate 3000型高效液相色谱仪(包括DAD检测器,赛默飞世尔公司);XSE205型电子天平(梅特勒-托利多公司,精度为十万分之一);ULUP-Ⅱ-10T型优普系列超纯水器(成都超纯科技有限公司)。
1.2 试药
甘草苷对照品(批号为111610-201607,含量为93.1%),甘草酸铵对照品(批号为110731-201720,含量为97.7%),橙皮苷对照品(批号为110721-201818,含量为96.2%),均购自中国食品药品检定研究院;甘草酸单铵盐A原料药(批号为1707003,含量为67.2%,北京凯因科技股份有限公司);复方甘草口服溶液(四川峨嵋山药业股份有限公司,批号分别为20160701,20160702,20160703,20160704,20160705,20160706);乙腈为色谱纯,磷酸、甲醇、三乙胺均为分析纯;复方樟脑酊、愈创甘油醚、甘油、浓氨溶液,均购自四川峨嵋山药业股份有限公司。
2 方法与结果
2.1 溶液制备
对照品溶液:称取甘草苷对照品17.84 mg,置100 mL容量瓶中,加80%甲醇溶解,并定容至100 mL,作为贮备溶液A;称取橙皮苷对照品64.12 mg,置250 mL容量瓶中,加甲醇溶解,并定容至250 mL,作为贮备溶液B;称取甘草酸铵对照品11.08 mg,置10 mL容量瓶中,加80%甲醇溶解,并定容至10 mL,作为贮备溶液C。分别量取贮备溶液A和贮备溶液B各1.00 mL、贮备溶液C 2.00 mL,置10 mL容量瓶中,加80%甲醇溶液,并定容至10 mL,作为混合对照品溶液。
供试品溶液:取样品(批号为20160701)4mL,置50mL容量瓶中,加80%甲醇溶液,并定容至50 mL,即得。
样品-橙皮苷混合溶液:另取样品(批号为20160701)4 mL,置50 mL容量瓶中,加入贮备溶液B 5.00 mL,用80%甲醇定容至50 mL,即得。
阴性对照品溶液:取复方樟脑酊18 mL、愈创甘油醚0.5 g、甘油12 mL、浓氨溶液适量,加水至100 mL,摇匀,作为阴性样品溶液;取上述溶液4 mL,置50 mL容量瓶中,加80%甲醇定容至50 mL,作为阴性对照品溶液;以80%甲醇作为空白溶液。
2.2 测定波长选择
提取混合对照品溶液和样品-橙皮苷混合溶液色谱图中甘草酸铵、甘草苷、橙皮苷色谱峰的紫外光谱图,各组分在252 nm(甘草酸铵)、276 nm(甘草苷)、284 nm(橙皮苷)波长处有最大吸收(见图1),故选择252,276,284 nm分别作为甘草酸铵、甘草苷、橙皮苷的测定波长。
2.3 色谱条件
色谱柱:Welch Ultimate C18柱(250 mm×4.6 mm,5μm);流动相:0.1%三乙胺(A,用磷酸调节pH至2.5),流动相B为乙腈,梯度洗脱(洗脱程序见表1);检测波长:252 nm(甘草酸铵)、276 nm(甘草苷)、284 nm(橙皮苷);流速:1.0 mL/min;柱温:30℃;进样量:10μL。
2.4 方法学考察

图1紫外光谱图

表1流动相梯度洗脱程序(%)
专属性试验:取2.1项下空白溶液、阴性对照品溶液、混合对照品溶液、供试品溶液、样品-橙皮苷混合溶液各10μL,按拟订色谱条件分别注入色谱仪中,测定,色谱图见图2。在284,276,252 nm为检查波长的色谱图中,混合对照品溶液各组分主峰保留时间相同的位置上,样品-橙皮苷混合溶液有相应色谱峰,各色谱峰分离度均大于1.5;供试品溶液有与混合对照品溶液甘草酸铵和甘草苷保留时间一致的色谱峰,而无与混合对照品溶液橙皮苷保留时间一致的色谱峰;阴性对照品溶液和空白溶液均无在与混合溶液保留时间一致的色谱峰。与混合对照品溶液保留时间一致的样品-橙皮苷混合溶液色谱图中,甘草酸铵、甘草苷、橙皮苷紫外光谱最大吸收波长分别为252,276,284 nm。样品-橙皮苷混合溶液色谱图中,匹配因子分别为甘草酸铵999.928/1 000,甘草苷999.911/1 000,橙皮苷999.954/1 000。
精密度试验:取2.1项下混合对照品溶液,按拟订色谱条件重复进样6次,以每次甘草酸铵、橙皮苷、甘草苷测定的峰面积计算。结果的RSD分别为0.35%,0.24%,0.31%(n=6),表明仪器精密度良好。
重复性试验:精密量取样品(批号为20160701)4 mL,依法制备供试品溶液共6份,按拟订色谱条件进样。结果甘草酸铵、橙皮苷、甘草苷峰面积的RSD分别为0.68%,0.22%,0.70%(n=6),表明方法重复性好。
稳定性试验:取重复性试验项下制备的供试品溶液10μL,分别于室温下放置0,2,4,6,8,12,24 h时,按拟订色谱条件进样测定。结果甘草酸铵、橙皮苷、甘草苷峰面积的RSD分别为0.28%,0.33%,0.26%(n=7),表明供试品溶液在室温放置24 h内稳定。

图2高效液相色谱图
加样回收试验:称取甘草酸单铵盐A(批号为1707003,含量为67.2%)334.82 mg,置100 mL容量瓶中,用80%甲醇溶解并定容至100 mL,作为甘草酸单铵盐A贮备液;取样品(批号为20160701,每1 mL样品中含甘草苷0.162 8 mg、甘草酸铵2.893 1 mg)2 mL,置50 mL容量瓶中,精密加入贮备溶液A 2 mL、甘草酸单铵盐A贮备液2 mL、贮备溶液B 5 mL,用80%甲醇定容至50 mL,得供试品溶液,平行制备6份。取10μL,按拟订色谱条件进样分析,按Cm=(Am×Cs)/(f×As)计算,Cm(μg)为回收率供试品溶液中甘草酸铵或甘草苷进样量,Am为回收率供试品溶液中甘草酸铵或甘草苷的峰面积,Cs(μg)为回收率供试品溶液中橙皮苷进样量,Cs为回收率供试品溶液中橙皮苷峰面积。结果见表2。
2.5 相对校正因子(f)值测定
取2.1项下混合对照品溶液2,5,10,15,20,25,30μL按拟订色谱条件分别注入色谱仪,校正截距为0,以进样量(m,μg)为横坐标、峰面积(A)为纵坐标进行线性回归,以甘草酸铵和甘草苷曲线斜率分别除以橙皮苷曲线斜率计算f值,并以3台不同液相色谱仪测定的平均值作为相对校正因子。结果甘草酸铵、橙皮苷、甘草苷线性回归方程分别为A=803.233 3m,A=1 786.872 0m,A=1 932.570 7m,r分别为0.999 3,0.999 2,0.999 5;线 性 范 围 分 别 为0.443 0~6.495 1μg,0.049 3~0.740 2μg,0.033 2~0.498 3μg。甘草苷相对校正因子fA为1.081 4(RSD=0.25%,n=3),甘草酸铵相对校正因子fB为0.449 2(RSD=0.22%,n=3)。
2.6 相对校正因子耐受性与相对保留时间(tR)考察
取2.1项下混合对照品溶液,以3台不同高效液相色谱仪、不同柱温(31.0,30.0,29.0℃)、不同测定波长(282/274/250 nm,283/275/251 nm,284/276/252 nm,285/277/253nm,286/278/254nm)、不同流动相pH(2.9,2.7,2.5)、不同流速(1.005,1.000,0.995 mL/min)的条件进行测定,按2.5项下方法计算f值,以及橙皮苷色谱峰分别与甘草苷色谱峰和甘草酸铵色谱峰保留时间的比值。结果见表3,表明不同液相色谱仪、柱温流动相pH、流速对tR无显著影响。
2.7 紫外光谱稳定性考察
取2.1项下混合对照品溶液,按2.6项下方法(除测定波长外)考察,提取橙皮苷、甘草酸铵、甘草苷紫外光谱图。结果橙皮苷在(284±2)nm、甘草酸铵在(252±2)nm、甘草苷在(276±2)nm波长处均有最大吸收;各成分紫外光谱图与各自对照品建立的紫外图库相比较,匹配因子均大于999.9/1 000。
2.8 样品含量测定
取样品4 mL,按2.1项下方法制备样品-橙皮苷混合溶液和供试品溶液。取样品-橙皮苷混合溶液和供品试溶液10μL,按2.3项下色谱条件,进样测定,记录色谱图,并按回收试验项下公式计算甘草酸铵和甘草苷进样量,通过进样量计算样品中甘草酸和甘草苷含量,测定结果采用配对t检验,与外标法测定数据无显著差异(P>0.05)。结果见表4。
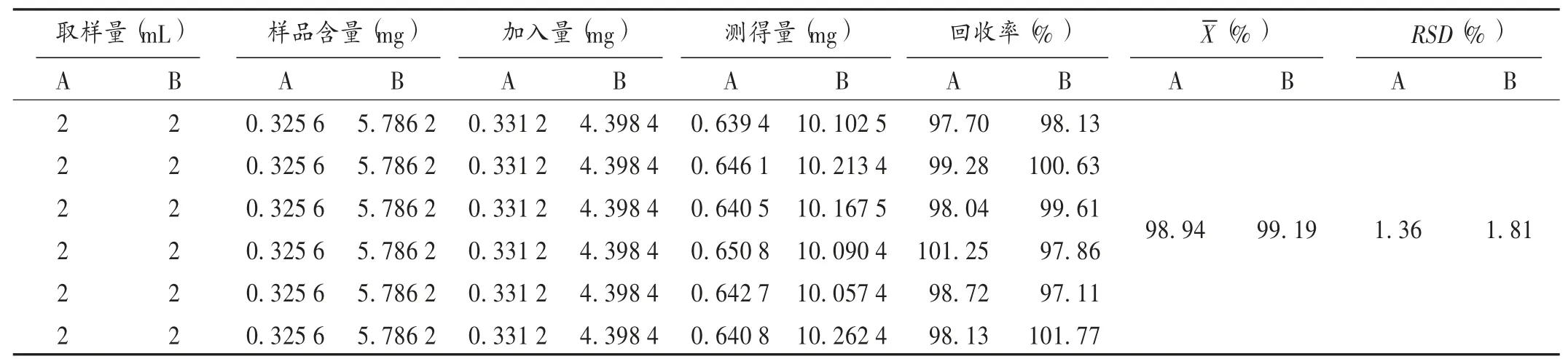
表2加样回收试验结果(n=6)
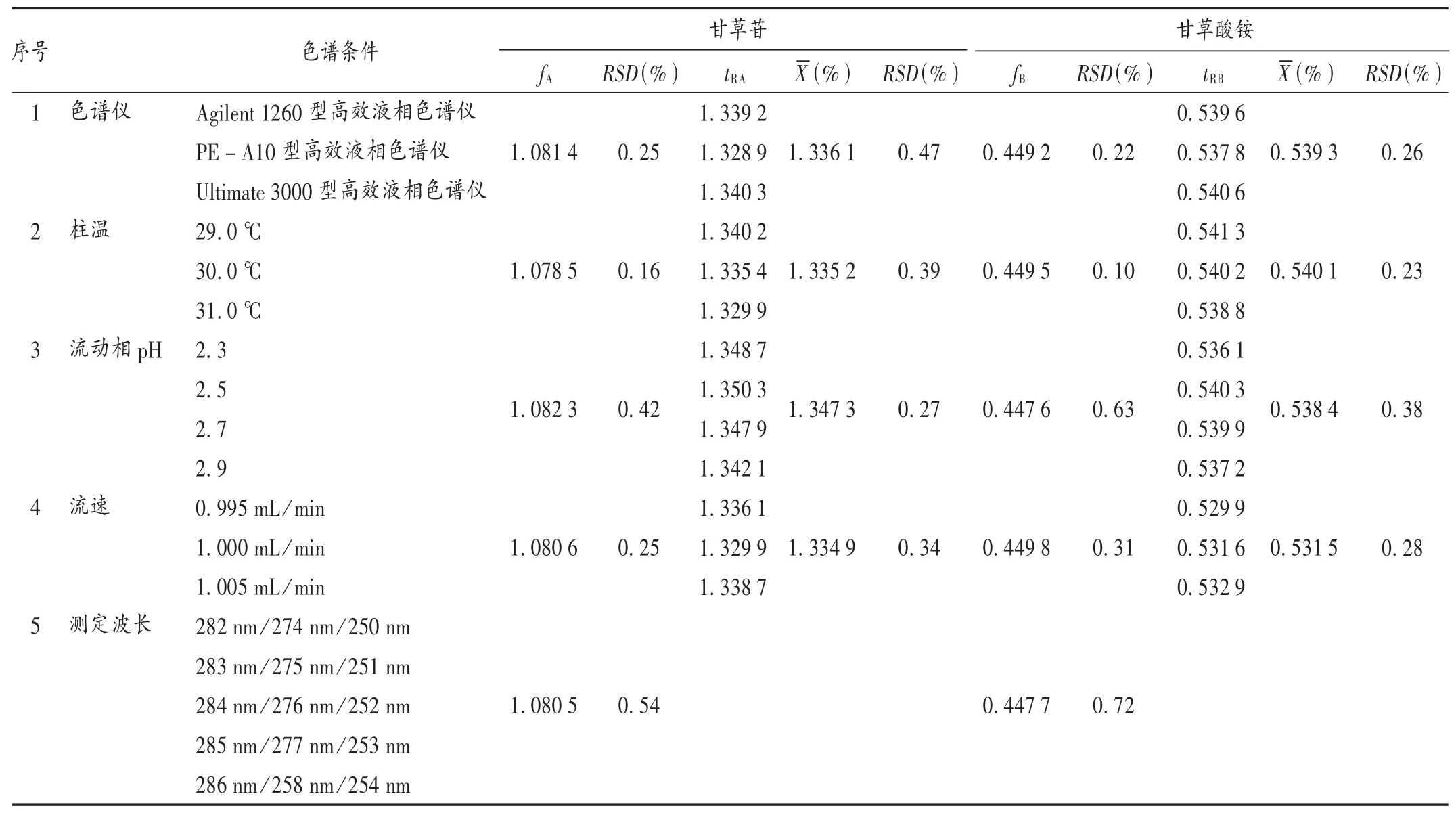
表3甘草苷和甘草酸铵相对校正因子耐受性试验和相对保留时间考察结果
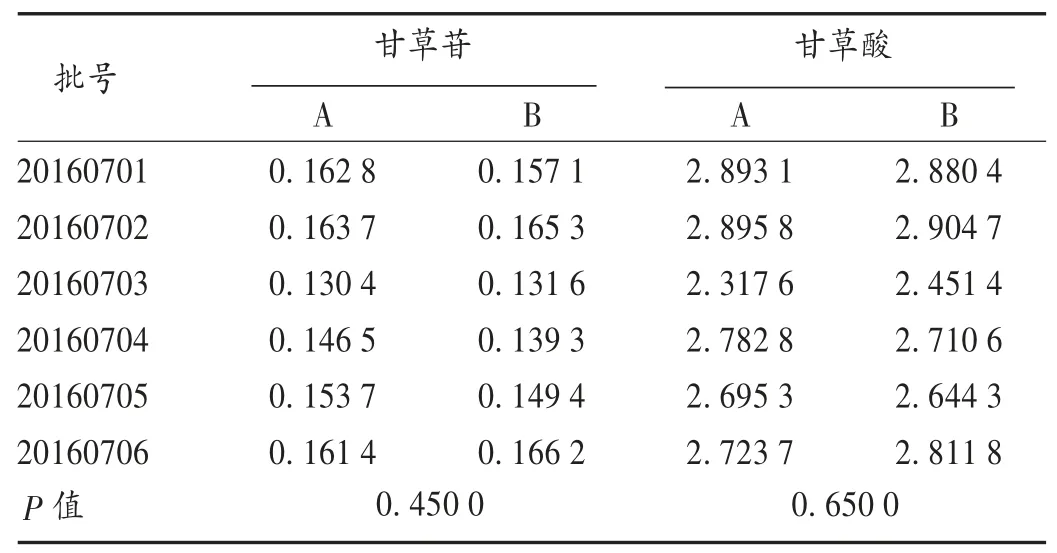
表4样品含量测定结果(mg/mL)
3 讨论
供试品溶液中定量加入替代对照品作为供试品溶液,替代对照品测定波长按试验设计的色谱条件,在未加入替代对照品的供试品溶液的色谱图中,不应有与替代对照品溶液保留时间一致的色谱峰。
由于供试品溶液杂质成分较多,对色谱柱有较高要求,未考察不同色谱柱相对保留时间的影响。参考国家药品监督管理局发布的国家药品标准YBH06492018,建议采用Welch Ultimate C18柱(250 mm×4.6 mm,5μm),本试验中采用以相对保留时间为辅助,各组分色谱峰提取紫外光谱图和对照品色谱峰所建立的紫外纯度检测图库比较为主的定性方法。由于工作站所建立的图库只能应用于同类色谱仪,故使用局限性较大。
替代对照品内加入法不需要单独制备与测定对照品溶液,可缩短试验时间,简化检验程序。可同时进样测定待测组分和对照品,可降低仪器噪音和操作过程的影响,结果准确,相对保留时间稳定。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中国药业(2021年12期)2021-06-28
中国应急管理科学(2021年4期)2021-04-13
中国合理用药探索(2021年2期)2021-01-03
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
中国中医急症(2019年10期)2019-05-21
考试周刊(2018年68期)2018-09-17
幸福·健康版(2018年3期)2018-03-23
食品与健康(2017年9期)2017-09-13
