
脊髓性肌萎缩伴呼吸窘迫1型1例报告
2020-12-03 07:07郑安洁郝丽红石武娟
临床儿科杂志 2020年11期
郑安洁 郝丽红 石武娟 李 征
天津市儿童医院新生儿内科(天津 300074)
脊髓性肌萎缩伴呼吸窘迫1 型(spinal muscular atrophy with respiratory distress type 1,SMARD1)是一种罕见的严重的常染色体隐性遗传神经肌肉疾病。患儿多宫内生长受限,婴儿期发病,以四肢肌力低下,下肢比上肢重,远端比近端重,以及生后6 周至6 月龄突然出现膈肌麻痹并呼吸窘迫、呼吸衰竭为主要表现[1]。SMARD1名称、临床表现与脊肌萎缩症有相似点,但发病基因、机制及临床表现有明显区别,临床上对SMARD1认识不足,极易漏诊、误诊。现报告分析1例SMARD1患儿的临床表现、神经电生理及基因检测结果,以提高对SMARD 1 的认知,争取早期识别,在已知存在患病风险的情况下对患儿父母做好遗传学指导和产前筛查。
1 临床资料
患儿,男,33日龄,因生后哭声弱,加重伴吃奶差2天于新生儿内科住院治疗。患儿系G2P2,因母扁平骨盆,胎儿生长受限38+5周行剖宫产。患儿出生体质量2 040 g,无窒息史,生后即哭,哭声弱,Apgar评分不详。生后因新生儿吸入性肺炎于当地住院治疗1周好转出院。患儿生后混合喂养,吸吮力欠佳,吃奶时间长(约30 min),两次吃奶间隔延长(约5~6小时),每次吃奶量无明显下降,吃奶时偶有呼吸急促、喉中痰鸣、面部微绀,吃奶后自行缓解。发病以来精神弱,睡眠时间增多(21~22 h/d)。患儿母亲妹妹的儿子因先天性胆道闭锁于8 月龄死亡。入院体格检查:体质量2 650 g,肛温36.7 ℃,呼吸52次/min,脉搏142次/min,收缩压、舒张压(SBP/DBP)75/38 mmHg,TcSO298%,神清,精神弱,哭声低弱,呼吸稍促,三凹征(-),无发绀,前囟平软,张力不高,颈软;双肺呼吸音粗,未闻及啰音;心音有力,心律齐,心率142次/min,心 前区未闻及明显杂音;腹软,未见肠型,肠鸣音存在,肝脏肋下1 cm,质软边锐;脐部未见渗出;睾丸已降至阴囊;四肢自发活动少,肌力正常,肌张力未见明显下降,双跟膝腱反射(+),双侧跖反射屈性,拥抱反射(±),吸吮反射(±),握持反 射 (+),觅食反射(+),四肢皮肤稍苍白,略发花,末梢温,脉搏有力,前臂内侧毛细血管再充盈时间2 秒。实验室检查:血糖5.52 mmol/L;血常规白细胞15.78×109/L,中性粒细胞32.6%,淋巴细胞51.4%,单核细胞10.5%,血红蛋白146 g/L,血小板190×109/L,C反应蛋白(CRP)<2.5 mg/L;血气分析、电解质、肾功能未见异常;谷氨酸氨基转移酶89~209 U/L,γ-谷氨酰转肽酶 135~232 U/L,天门冬氨酸氨基转移酶 78~383 U/L;肌酸激酶 527 U/L,肌酸激酶同工酶 42 U/L;促甲状腺激素 15.064 mIU/L;血氨、血尿代谢病筛查未见异常。腰椎穿刺脑脊液检查无异常。胸腹联合摄片:双肺纹理重,心膈无著变,腹部未见明显异常。心电图、超声心动图、腹B超检查未见异常。
入院后予鼻饲喂养、呼吸道管理、抗感染,维持内环境稳定,加用优甲乐及对症支持治疗,住院3天间断出现呼吸喘促、吸气性喉鸣、三凹征(+)伴心率增快、发绀、血氧饱和度下降,予头罩吸氧、侧卧位好转,多于鼻饲奶后1小时左右出现,住院5天行喉CT平扫未见异常,肺CT平扫提示双肺散在小片状炎性实变;双侧胸膜增厚,喉、肺CT三维重建未见异常;神经电生理(住院3天)第一次检查四肢肌电图未见异常。
住院第9 天,患儿出现呼吸衰竭,血气分析pH值7.138,PCO2>110 mmHg,PO257.3 mmHg,SO289.2%,予气管插管、呼吸机使用下仍间断出现自主呼吸喘促、三凹征(+)伴发绀、血氧饱和度下降,发作当时心率170~180 次/min,右肺呼吸音消失,调整呼吸机参数或吸痰,尤以改变体位为左侧斜卧位时能很快缓解症状。气管插管后床旁胸片示右侧膈膨升及炎性实变(图1)。患儿发作性呼吸困难伴吸气性喉鸣,不除外气管或支气管软化,但因家属拒绝,未行纤维支气管镜检查证实。
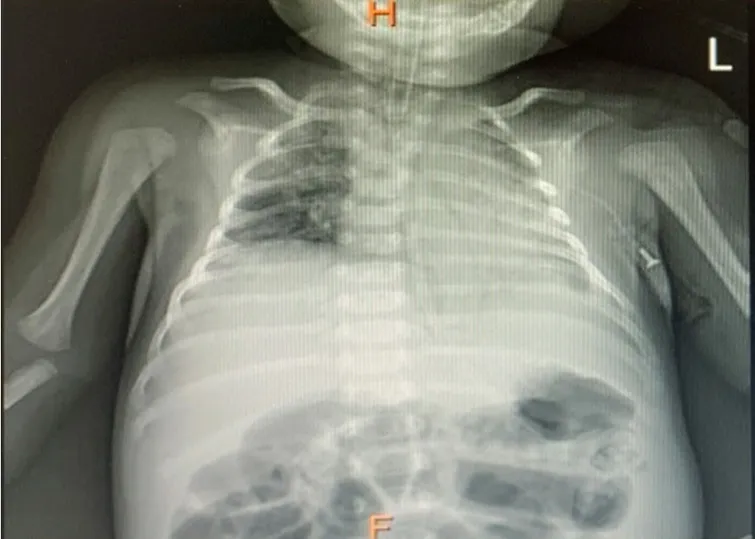
图1 患儿第2 次胸片
经家属知情同意完善染色体核型分析:46,XY,正常。送天津金域医学检验实验室进行复杂疾病基因检测结果发现IGHMBP 2基因两个杂合的致病变异,母亲来源无义突变c.1813 C>T,父亲来源缺失突变c.905_912+84del。
住院第19天,第二次神经电生理检查神经电图示双上肢正中神经运动神经传导速度、尺神经运动神经传导速度复合肌肉动作电位缺失,双下肢胫神经运动神经传导速度远端复合肌肉动作电位可引出,其潜期延长、波幅降低,近端复合肌肉动作电位缺失,提示传导阻滞,双下肢股神经潜伏时检测大致正常;双上肢正中神经感觉神经传导速度、尺神经感觉神经传导速度及双下肢胫后神经感觉神经传导速度缺失;肌电图:所检肌运动单位电位时限、振幅及募集反应正常,未见自发电位;提示多发性周围神经损害。脑干电测听示双耳Ⅰ、Ⅲ、Ⅴ波可引出,左侧各波潜期轻度延长,各间期正常,左耳听力轻度下降;右侧各波潜期及间期正常,右耳听力正常;提示左侧外周段轻度异常,左耳听力轻度下降。
住院20 天,家属放弃治疗,拔管出院,出院后数小时患儿死亡。
2 讨论
SMARD 1 是一种罕见的致命性疾病,位于常染色体11q13.3的IGHMBP2基因发生变异,主要累及脊髓前角α 运动神经元,在生后6 周至6 月龄内因膈肌 麻痹发生呼吸衰竭和引起严重的婴儿轴突神经病变[2]。基因检测结合临床表现可诊断SMARD1。
IGHMBP 2编码的DNA/RNA 螺旋酶包含一个DNA 螺旋酶结构域(氨基酸19-641),一个单链核酸结合的R3H基序(氨基酸726-784),以及一个锌指区(氨基酸897-940)[2]。该螺旋酶催化双链DNA解旋,参与RNA合成,在细胞核中参与调控转录及tRNA的加工[3]。研究发现IGHMBP2的突变分布在除R3H基序外的整个蛋白质中,并且主要位于螺旋酶结构域[2],以点突变为主,亦有内含子倒位、大片段缺失[1]。
本例患儿以膈肌麻痹、呼吸衰竭为表现,基因检测发现2 个杂合的致病变异,无义突变c.1813 C>T为母亲来源,IGHMBP 211 q 13.3 NM_002180.2 Exon13 c.1813C>T p.(Arg605*),该变异为无义突变(预计会使所编码的蛋白质第605 位氨基酸由Arg变为终止密码子,并使得蛋白质翻译提前终止)。该变异预计会导致所编码的蛋白质发生截短从而丧失其正常功能。有文献报道在脊肌萎缩症伴呼吸窘迫1 型患者中检测到该变异[4];ESP 6500 siv 2_ALL、千人基因组(1000 g 2015 aug_ALL)和dbSNP 147 数据库均未见收录。缺失变异c.905_912+84 del 为父亲来源,IGHMBP 211 q 13.3 NM_002180.2 Exon 6 c.905_912+84 del p.(Gln 302 fs),该变异为内含子与外显子交界处的缺失变异(预计会导致所编码的蛋白质自第302 位氨基酸Gln 开始发生移码,并导致翻译提前终止)。该变异预计会导致所编码的蛋白质发生截短从而丧失其正常功能。HGMD 数据库未见文献报道该变异;ESP 6500 siv 2_ALL、千人基因组 (1000g2015aug_ALL)和dbSNP147数据库均未见收录。
SMARD1发病主要影响脊髓前角α运动神经元,其周围神经病理表现为轴突变性,肌电图显示广泛神经源性损伤,感觉神经传导多数被保留[5]。本例患儿自发病至死亡肌力及肌张力无明显下降,第1 次四肢肌电图检测未见异常,第2 次神经电生理检查以多发性周围神经损害为表现,脊髓前角未见病变,周围神经运动及感觉神经均受到累及,且上肢重于下肢,与既往所述不完全一致。有报告1例合并甲状腺功能减退、先天性心脏病和小头畸形的SMARD1患儿,其神经传导研究提示感觉运动神经均受累及[6]。本例患儿除表现感觉运动神经损伤,同样有甲状腺功能异常。近年发现,IGHMBP 2也是2 型Charcot-Marie-Tooth病(CMT 2)的致病基因。CMT 2 的周围神经病理也表现为轴突变性,纤维末梢和淋巴母细胞研究表明,CMT2的IGHMBP2蛋白水平高于SMARD1,提示临床表型和蛋白水平之间存在关系[7]。研究发现,408例疑似CMT病或其他遗传性外周神经病(IPNs)患者中发现4 例IGHMBP 2纯合或复合杂合变异者,其中3 例表现为儿童轴突主导的感觉运动多发性神经病,而另1 例被诊断为SMARD 1,表现为低出生体质量,哭声微弱,自发活动减少,生后4个月出现呼吸窘迫。目前,没有证据表明SMARD 1 或CMT 2 患者在遗传水平上是可区分的,而蛋白质水平的变异与临床严重程度之间的负相关仍存在争议[2]。同时,本例患儿还表现有肝功能损害、心肌酶异常,呼吸困难发作时心率加快,可能影响自主神经功能,亦不除外气管支气管 软化症。说明SMARD 1 临床表型存在较大的差异性。此外,本例患儿发作时与进食、体位的关系及查体一侧呼吸音消失等特点,也需临床仔细观察及进一步思考。
基因变异的多态性使得疾病临床表型及严重程度不一,疾病诊断在临床表现基础上结合基因检测的优势,在精准医疗服务的同时可提高对疾病的认知,基因检测做为诊断的一个辅助手段需合理应用。
猜你喜欢
心电与循环(2021年4期)2021-11-29
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
小学科学(学生版)(2019年10期)2019-11-16
中学数学杂志(2019年9期)2019-05-29
中国外汇(2019年23期)2019-05-25
中国环境监察(2017年5期)2017-10-23
中国眼镜科技杂志(2016年17期)2016-10-24
百科知识(2015年18期)2015-09-10
中国当代医药(2015年30期)2015-03-01
