
巨脑回畸形并皮层下带状灰质异位1例临床及基因变异分析
2020-12-03 07:07:46张鑫阳周福军崔清洋
临床儿科杂志 2020年11期
张鑫阳 周福军 崔清洋
新乡医学院第一附属医院儿科(河南卫辉 453100)
无脑回畸形(Lissencephaly,LIS;孟德尔遗传,编号607432,257320,611603,614019分别对应于Ⅰ型至Ⅳ型;300067 和300215 分别对应于X 连锁Ⅰ型和Ⅱ型)是一系列罕见的由神经元迁移缺陷导致的皮层发育畸形,估计发病率为0.12/万[1]。LIS谱系包括无脑回(脑沟间距> 3 cm的大脑皮层区域)、巨脑回(脑沟间距1.5~3.0 cm 的异常宽脑回)和皮层下带状灰质异位(subcortical band heterotopia,SBH,位于大脑皮层深部并由一层薄的白质隔开的纵向灰质带)[2]。
一般认为巨脑回是无脑回的特殊类型,二者为同类神经细胞移行和皮质构成异常的不同程度。巨脑回与无脑回畸形可合并存在,且其临床症状一般较单纯性巨脑回畸形更严重[3]。所有LIS患者均有智力障碍,但LIS 亚型不同,其严重程度也存在显著差异。临床表现为从重度残疾和短生存期的无脑回,到轻度智力或学习障碍的部分性SBH。无脑回畸形儿童在出生后的前几周至几个月内表现为神经功能缺陷,包括吃奶少、轻度肌张力减退、异常弓背行为或角弓反张。在生命的第一年,发育落后或癫痫发作活动也很常见[4]。
现回顾分析1例巨脑回畸形并皮层下带状灰质异位患儿的临床及基因检测结果。
1 临床资料
患儿,男,1 个月25 天,傈僳族,因发现逗引反应差、睡眠多1周入院。入院1周前发现患儿逗引反应差,睡眠多,双眼不跟踪移动物体,偶伴轻微肢体抖动,持续约2秒,无双眼上翻凝视、口吐泡沫及意识丧失。患儿系G3P1,前2胎均为自然流产(原因不详)。患儿生后第3天出现黄疸,持续时间长,口服药物好转(具体不详)。父母体健,非近亲结婚,母孕期无异常,家族中无类似病史。入院体格检查:体温36.6℃,体质量6.5 Kg,脉搏123次/min,呼吸35次/min,头围38 cm,神清,反应差,前囟平软(1.5 cm×1.5 cm),心、肺、腹未见异常,双下肢肌张力稍高,四肢肌力4级,觅食反射、吸吮反射均可引出,拥抱反射、握持反射减弱,双侧膝腱反射可引出,双侧巴氏征对称阳性。脑电图正常。头颅磁共振成像(MRI)示双侧额顶枕叶脑回体积增大,边缘平直、脑沟明显减少,灰白质比例不协调;脑白质明显减少,脑灰质增厚、增多;第三脑室及双侧侧脑室稍扩大,胼胝体较细;透明隔间腔稍宽;考虑双侧额顶枕叶巨脑回畸形并皮层下带状灰质异位(图1)。0~6岁小儿神经心理发育评估,发育商25,全面发育迟滞。
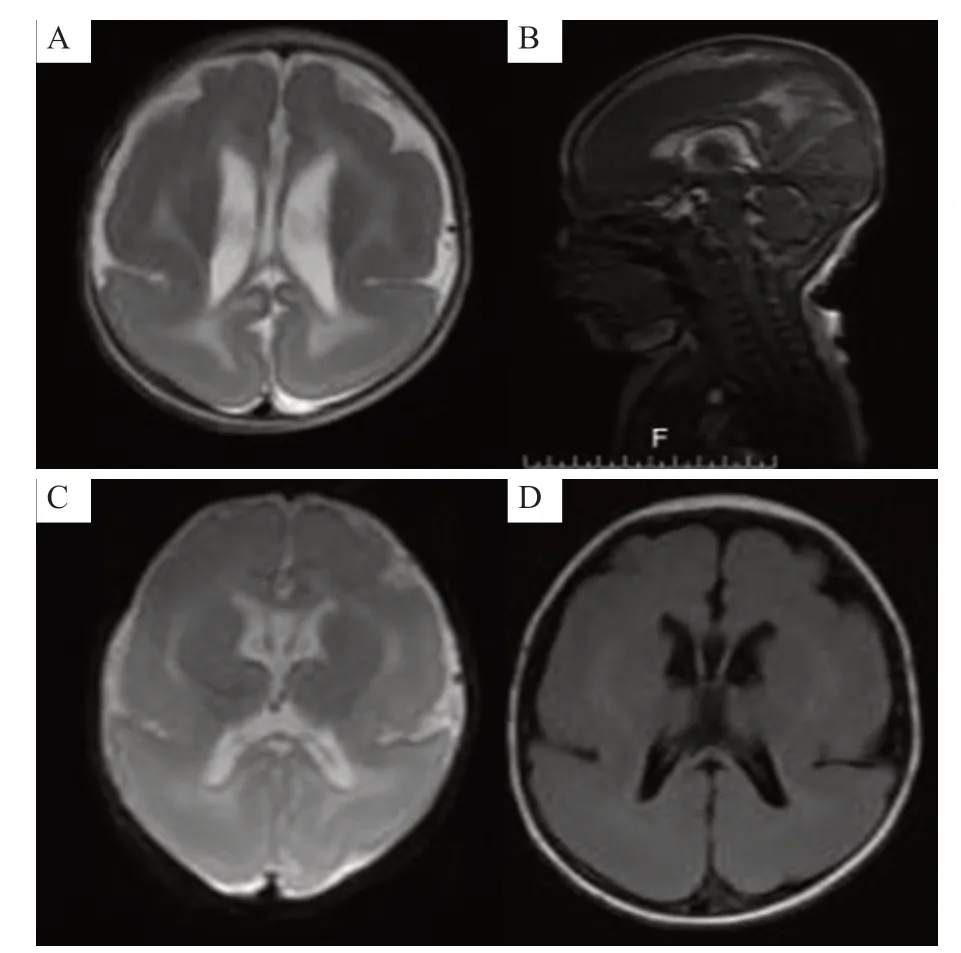
图1 患儿头颅MRI 表现
经家长知情同意后,采集患儿及其父母外周血2 mL,乙二胺四乙酸(EDTA)抗凝,由北京康旭医学检验有限公司进行基因检测。DCX基因(exon3)正向引物序列:TGA TTC ATT GCT TTG CCT GCT,反向引物序列:CAC AGG CCA GGG AGA ACA AG;PCR扩增条件为:95 ℃预变性10 min;95 ℃变性30 s(35个循环);60 ℃退火 30 s(35 个循环);72 ℃延伸45 s(35 个循环);72 ℃彻底延伸 5 min(35 个循环)。结果发现患儿DCX基因c.829 C>T 的核苷酸变异,该变异导致第277 号氨基酸由精氨酸转变为半胱氨酸(p.Arg 277 Cys),为错义变异。c.829 C>T 变异位点SIFT 软件预测为Deleterious,Polyphen 2 软件预测为Probably damaging,MutationTaster软件预测为disease causing。家系验证发现患儿父亲未携带该位点变异,母亲为该变异位点杂合子(图2)。
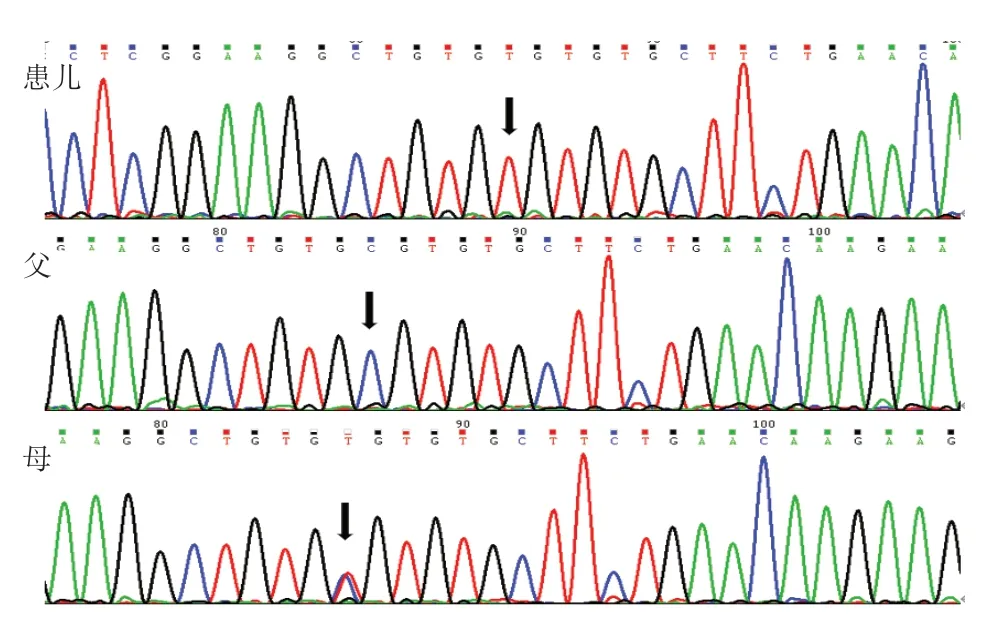
图2 DCX 基因c.829C>T 变异测序图
根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)联合美国分子病理学会(Association for Molecular Pathology,AMP)2015 年制订的“基因序列变异的解释标准和指南”进行致病性分析[6]。判断为疑似致病变异。结合患儿头颅MRI、临床表现确诊巨脑回畸形并皮层下灰质异位。
患儿间断进行康复治疗,1岁半时再次进行“0~6岁小儿神经心理发育评估”,发育商41,仍有全面发育迟滞;之后不久患儿癫痫大发作1 次,建议来院进行24小时动态脑电图检查,家属拒绝。
2 讨论
80%的巨脑回畸形的发生与LIS1基因和DCX基因相关,其中DCX基因突变常和性别相关联,男性患儿常表现为无脑回畸形,而女性患儿常表现为双侧皮层下带状灰质异位[5]。女性中与DCX基因相关的SBH高发生率可用莱昂作用解释。X染色体失活在发育中的女性杂合子大脑中产生两个细胞群,即携带失活的缺陷DCX等位基因神经母细胞迁移正常,而携带失活的典型DCX 等位基因神经母细胞因迁移不足形成异位带。在半合子男性中,有缺陷的DCX等位基因将被激活而导致更严重的表型。罹患典型DCX 相关无脑回畸形的男性通常早期出现严重的认知、语言障碍和癫痫发作等症状[6]。
神经元迁移是脑发育过程中的重要阶段,其过程受理化因素与生物性损害及遗传因素影响后均可导致生成的神经细胞从胚胎生发基质向大脑表面移行过程受阻,造成神经元移行异常,包括脑裂畸形、灰质异位、无脑回、巨脑回及多微脑回等。正常的大脑皮层分为六层,即分子层、外颗粒层、外椎体细胞层、内颗粒层、内椎体细胞层和多形细胞层,而DCX基因相关的无脑回或巨脑回畸形仅有模糊的四层,即分子层、浅表细胞层、细胞稀疏层、深部细胞层,与PAFAH1B1基因相关无脑回畸形相似,但II 层相对较薄(锥体细胞)。在正常皮层中I层对应的是巨脑回或无脑回畸形的分子层,巨脑回或无脑回浅层对应正常脑皮层II-IV层,细胞稀疏层对应皮质下白质层,深部细胞层对应灰质异位层的未分层的异位神经元。
大脑的皮质发育畸形与Reelin通路有关[8]。Reelin在细胞膜上与ApoER 2、极低密度脂蛋白受体(very low density lipoprotein receptor,VLDLR)及α3β1整合蛋白结合,使下游胞质内的衔接蛋白disabled 1(Dabl)募集,募集的Dabl 进一步磷酸化Sre 酪氨酸激酶家族中的Sre 和Fyn 激酶,磷酸化的Dabl 对 Fyn和Src 具有正反馈效应引起其进一步激活,进而可进一步磷酸化细胞周期依赖性激酶5(cyclin-dependent kinase 5,Cdk5)激活微管相关蛋白即双皮层蛋白而作用于神经元的微管蛋白和肌动蛋白,影响神经元的定位[9]。且Reelin 具有细胞支架蛋白的作用,可调节皮层中放射状胶质细胞纤维的排列,确保平行排列的纤维为神经元的正确迁移提供支架,Reelin -Dabl 信号可 能是神经元迁徙的起始或终止信号,保证大脑新皮质的发育遵从从内到外的发育模式,形成正常的大脑皮层[10]。
DCX基因在Reelin通路中发挥重要作用,对神经元的迁移意义重大[11]。DCX基因定位于Xq22.3-q23,全长117.37 kb,包括7 个外显子及6 个内含子,其编码基因长为9405 bp,编码441氨基酸。DCX基因编码双皮层蛋白,在发育中的大脑中以多种剪接变异体的形式表达,尤其是在侧脑室的亚带(Subventricular zone,SVZ)和齿状回颗粒细胞层下区(subgranular zone,SGZ)迁移细胞中。DCX富集在神经突触远端。DCX 是一种微管相关蛋白,与微管结合有助于微管稳定和组装[12]。双皮层蛋白包含2 个由80 个氨基酸组成的高度重复保守而有序的微管蛋白结合域(即“N-DC”DCX蛋白的46-139位氨基酸和“C-DC”173-263位氨基),该区域是调节蛋白磷酸化及蛋白质间相互作用所必需的,在神经细胞移行过程中促进微管蛋白网络结构的延伸及稳定,该蛋白的磷酸化可影响它与微管蛋白的相互作用,许多文献报道DCX基因相关的无脑回畸形变异往往集中在这两个区域[13-14]。研究发现,C-DC结构域错义变异引起的临床症状比N-DC结构域错义变异的临床症状轻微[15]。DCX基因变异可造成双皮层蛋白结构改变,最终影响神经元移行过程。DCX基因突变可表现为无义变异或错义变异,变异可集中于2~7 外显子的任一部位,于第2 至第4 外显子的基因变异主要为错义变异,而位于第5 至第7外显子的变异类型主要为无义变异。本例患儿DCX基因c.829C>T位于第3外显子,为错义变异,支持文献报道[16]。
头颅MRI对于区分脑畸形发育具有重要作用[17]。参考国内分级标准,可将巨脑回的严重程度分为6级,严重程度依次递减,其中1级最重,6级最轻。1级,完全无脑回;2 级,无脑回,前部脑皮质有少量浅沟;3级,后部无脑回而前部为巨脑回;4级,后部重于前部的弥漫性巨脑回;5 级,巨脑回与皮层下带状灰质异位混合出现;6级,单纯性皮层下带状灰质异位[18]。多于90%的皮质下灰质异位症的患者为女性,其发病原因通常由DCX基因杂合变异引起,而且DCX基因相关的SBH 发生部位主要位于大脑额顶叶区域[19]。本例患儿的变异位点c.829C>T chrX:110644337在转录本NM_178153下表示为:c.586C>T,属于C-DC区域,曾有报道[20]。该基因变异不属于多态性变化,发生频率极低,在国内尚未见报道。结合患儿头颅MRI表现,参照何丽等[18]报道的分级标准,符合3 级,提示此位点变异可能引起严重的脑发育畸形。巨脑回与无脑回畸形常合并存在,且其临床症状一般较单纯性巨脑回畸形更严重,国内胡春辉等[21]报道巨脑回畸形合并无脑回畸形14例。另有11例巨脑回合并灰质异位,7例巨脑回畸形合并无脑回畸形的报道[22]。本例患儿头颅MRI提示后部为无脑回而前部为巨脑回,临床表现重度全面发育迟滞,支持文献报道。同时该患儿为傈僳族,其发生突变结果不同是否与种族有关,尚不清楚。
在与DCX基因相关的巨脑回畸形中接近10%由于自身基因变异引起,在女性中,通常表现为皮层下带状异位,在男性中常表现为经典的巨脑回畸形。对于DCX基因杂合的女性来说,后代中男孩发病率为50%[19]。本例患儿父亲DCX基因无异常,母亲为杂合子,无家族史,符合X 连锁遗传方式,后代男孩出现巨脑回概率为50%,可疑致病变异位点c.829C>T(P.Arg277Cys)对于产前诊断具有重要作用,对于优化人口素质有重要意义。本例患儿发育商低于正常范围,脑畸形较重,既往研究表明癫痫是该病的主要临床表现之一,本例患儿在1岁半后癫痫大发作1次,支持文献报道。
综上所述,本例患儿皮质下带状灰质异位合并巨脑回畸形,目前全面发育落后。许多文献报道LIS 可出现难治性癫痫[23],该患儿目前尚未发作,而且经过早期康复干预患儿病情好转,需长时间追踪病情变化。同时,DCX基因c.829C>T错义变异致病,国内尚未见报道,丰富了LIS基因库,并有助于遗传咨询和产前诊断。
猜你喜欢
科学导报(2023年68期)2023-10-04 22:42:06
中国现代医药杂志(2020年3期)2020-05-08 04:33:08
中国生物医学工程学报(2019年6期)2019-07-16 07:52:48
中国临床医学影像杂志(2019年2期)2019-04-25 06:15:38
中国临床医学影像杂志(2019年2期)2019-04-25 06:15:38
中外医疗(2016年15期)2016-12-01 04:25:39
婚姻与家庭·性情读本(2016年5期)2016-05-14 21:33:38
中国纺织(2015年2期)2015-03-16 23:41:58
中国CT和MRI杂志(2014年7期)2014-06-27 05:49:11
计算机工程(2014年6期)2014-02-28 01:26:26
