
新生儿原发性先天性淋巴水肿一家系基因分析并文献复习
2020-12-03 07:07:48高金枝
临床儿科杂志 2020年11期
魏 莹 高金枝 陈 玲
华中科技大学附属同济医院新生儿科(湖北武汉 430030)
先天性胎儿水肿是新生儿常见疾病,其病因多样,常见免疫性及非免疫性因素两大类[1]。免疫性先天性胎儿水肿主要指血型不合引起的溶血性疾病;非免疫性因素包括先天性心肺疾病、贫血、感染、染色体异常等。非免疫性因素中,先天性淋巴管系统发育缺陷所致的原发性淋巴水肿(primary lymphedema,PL)比较罕见,是由基因变异引起的遗传性疾病,包括PLⅠ和PLⅡ两种类型。PLⅠ也称为原发性先天性淋巴水肿(primary congenital lymphedema,PCL),以淋巴管缺失或减少为特征,临床上在出生时即有水肿。PLⅡ主要在青春期前后起病[2]。1892年首次报道PCL,也称为Milroy 病[3]。PCL 主要表现为双下肢的无痛性、非凹陷性水肿,可累及整条腿,也可局限于足部甚至脚趾,少数可累积胸腹腔。PCL 极其罕见,其发病率约1/6000,男女比例1∶2.3[4]。PCL属于常染色体显性遗传,目前其唯一已知的相关基因变异即血管内皮生长因子受体(vascular endothelial growth factor receptor-3,VEGFR3)基因(fms related receptor tyrosine kinase 4,FLT4),位于5q35.3[4]。目前为止,有关PCL 已有报道超过50 种FLT 4基因变异,其中C.2797G>C、C.2569G>A、C.3122G>A、C.3323 _3325delTCT等是目前被报道次数最多的变异位点[5]。Evan 等[6]曾报道分析了12 个PCL 家系,发现了8 个FLT 4 的变异位点,其中3 个均为上述常见的变异位点 。Connell 等[7]也曾分析了52 例原发性淋巴水肿的患者,发现有阳性家族史的患者中75%存在FLT 4相关的基因变异,无阳性家族史的患者中68%存在FLT 4相关的基因变异。本文回顾性分析华中科技大学同济医学院附属同济医院诊治的1 例PCL患儿的临床资料和基因变异特征,并综合文献进行分析。
1 临床资料
先证者,男,5天,G2P1,单胎,38周+4,出生体质量3 650 g,因母亲Rh阴性、贫血、乙肝表面抗原携带、胎儿右侧胸腔积液,催产后顺产出生。出生时无窒息,Apgar评分为9分、10分,羊水清,脐带、胎盘无异常。出生后因Rh 溶血高危儿、胸腔积液待查在外院住院治疗。
患儿曾在胎儿期,妊娠18 周时,彩超提示右侧胸 腔内见前后径约0.22 cm 的液性暗区,无双下肢水 肿。后定期监测,胸腔积液逐渐增多,妊娠29 周时,彩超示右侧胸腔见范围约1.5 cm×0.53cm液性暗区。患儿出生当天血红蛋白(Hb)160 g/L,总蛋白(TB)43.9 μmol/L,余血常规及生化检测未见明显异常。出生第5天,Hb 132 g/L,TB 221 μmol/L,余血常规及生化检测未见明显异常;彩色多普勒超声心动图未见异常;肺部彩超示右侧胸腔内见前后径约2.7cm液性暗区。曾在外院行溶血三项检测(抗体释放试验、游离抗体试验、直接抗人球蛋白试验)均为阴性。给予阿莫西林克拉维酸钾抗感染、营养心肌、脱脂奶(澳洲美可卓蓝妹子脱脂奶粉)喂养等治疗后,胸腔积液无改善。
为求进一步诊治转入华中科技大学附属同济医院。入院体格检查:体温37.8 ℃,呼吸43 次/min,脉搏146 次/min,收缩压/舒张压(SBP/DBP)70/38 mmHg,体质量3.78 kg,精神反应尚可,足月儿外貌,前囱平软,头部未及包块,全身皮肤中度黄染,颈软,未见吸气三凹征;双肺呼吸音粗,未闻及明显啰音,右下肺呼吸音稍减低;心腹无异常;脐轮无红肿;四肢肌张力无异常,双下肢非凹陷性水肿;原始反射正常引出。入院实验室检查:Hb 132 g/L,TB 221 μmol/L,余血常规、C反应蛋白、降钙素原及生化检测未见明显异常;甲状腺功能无异常;TORCH 及微小病毒B19、肺炎支原体、衣原体、地中海贫血等检查均无异常。肺部CT示右肺下叶实变不张,右侧胸腔积液。腹腔B 超未见积液。胸水检测示黄色浑浊,李凡他试验阳性,有核细胞计数 3 400×106/L(淋巴细胞 81%,中性粒细胞 7%),红细胞计数 680×106/L,乳糜试验阴性,培养阴性。出生后7 天查肺部彩超示右侧胸腔右上肺野肺实变伴支气管充气征、右侧胸腔积液(最大前后径2.3 cm),彩色多普勒超声心动图示卵 圆孔未闭(3 mm)、心包积液(2 mm)。出生第9天,Hb 131 g/L,TB 172 μmol/L,余血常规及生化检测未见明显异常。出生第21天,Hb 107 g/L,TB 41.8 μmol/L,余血常规及生化检测未见明显异常。
入院后起初继续脱脂奶喂养、头孢哌酮抗感染、补充静脉营养等治疗。直至生后12天,患儿体质量无增长;复查肺部彩超示右侧胸腔右上肺野肺实变伴支气管充气征、右侧胸腔积液(最大前后径2.2 cm);彩色多普勒超声心动图示卵圆孔未闭(3 mm)、心包积液(2 mm),胸腔积液及心包积液均无好转。遂改为普通配方奶喂养,停抗生素治疗。后患儿胸腔积液逐渐自行吸收减少,双下肢水肿逐渐局限于双足背部。出生第20 天,复查肺部彩超示右上肺野实变较前好转,右侧胸腔积液(最大前后径0.7 cm);彩色多普勒超声心动图示卵圆孔未闭(2.8 mm)、未见心包积液。患儿病情好转,遂予以办理出院。
追问病史,患儿父亲出生后即出现双下肢轻度水肿,未发现胸腔积液及其他疾病,后渐自愈。母亲健康,无特殊疾病史。非近亲婚配。母乳妊娠第1胎时发现胎儿全身水肿、大量胸腔积液及腹腔积液,故而引产。为查清病因,经医学伦理审核以及家长知情同意,采集患儿及其父母血各2 mL,EDTA抗凝管送至上海明码生物科技有限公司行基因分析。对患者基因组DNA进行全外显子组捕获和测序,基于二代测序数据进行点变异及小片段插入缺失变异(SNV)和大片段拷贝数变异(CNV)分析,并对候选变异位点在家系样本DNA 中进行Sanger 测序验证。结果患儿检测出FLT4基因杂合变异,c.3820 G>A(p.Asp1274Asn),经过Sanger测序验证,该变异来源于父亲,见图1。目前未见文献以及大规模人群频率数据库报道该位点突变,但已有大量文献报道FLT4基因的近50余种变异均可导致胎儿水肿的发生[3-5]。
患儿出院1 个月后我科进行电话随访,家属诉患儿于1 月龄时在外院复查胸腔彩超示未见胸腔积液,仍有轻度双下肢水肿。
2 讨论
本例患儿产检时即有双下肢水肿及胸腔积液,出生后溶血三项结果阴性、贫血不严重、彩色多普勒超声心动图未见严重的结构异常,相关病原学及炎症指标无阳性发现,抗感染及脱脂奶治疗无效,排除溶血、贫血、先天性心脏病及感染等病因,结合基因检测结果,考虑患儿为PCL诊断明确。
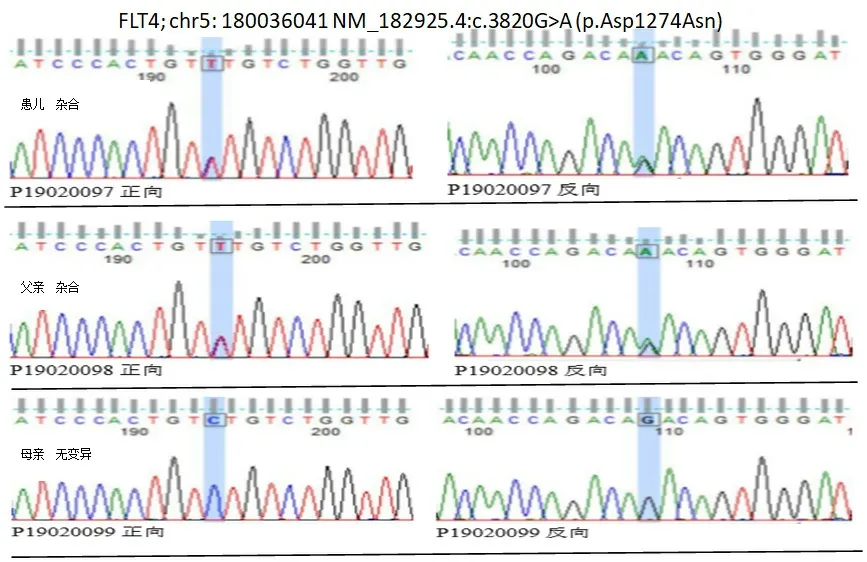
图1 患儿及父母的基因测序结果
PCL是一种罕见的家族性遗传病,大多数基因变异均为常染色体显性遗传,也有个别报道存在常染色体隐性遗传[2,5,8]。家族内同一基因变异类型的临床表现不同,同时不同的基因变异可导致相同的临床表现[6]。该病以下肢无痛性、非凹陷性水肿为特征,绝大部分在出生时(或出生前)或出生后不久即出现足部水肿,常呈双侧对称分布,偶有不对称分布。研究发现,该病是由于微淋巴管(尤其是肢体末端的局部淋巴管)先天性发育不全所致[4]。由于局部淋巴管发育不全,使淋巴回流受阻、淋巴内压升高、淋巴管曲张及瓣膜破坏,导致淋巴液滞留于组织间隙而发生水肿[9]。PCL 中的淋巴水肿为慢性良性病程,据报道,该病引起的水肿可终生存在,但偶有个例报道该水肿可自发消退[7,10]。该病除了受累部位的慢性不适外,不影响终身寿命,很少发生严重并发症,偶有合并肠淋巴管扩张、足背和脚趾的细菌感染、复发性感染性关节炎、血管肉瘤和淋巴管肉瘤等的个例报道[10]。
本研究家系中,未出生的第一胎及先证者均表现为胸腔积液及双下肢水肿,第一胎甚至出现大量腹水,但先证者父亲仅表现为自愈性的双下肢水肿,进一步证实该病临床表现即使在家族内同一基因变异类型中亦存在较大异质性。另外,经过随访观察,先证者的胸腔积液逐渐减少、自行吸收,仅有轻度双下肢水肿,也充分证实了该病为良性病程。
早在2 0 世纪末,便已有报道P C L 相关的基因位于染色体5 q 35[11-12]。该区域的基因FLT 4编码VEGFR-3,是目前唯一已知的相关变异基因。VEGFR-3 被认为参与了淋巴管生成和淋巴管内皮的维持过程[13]。FLT 4基因包含31 个外显子,其中30 a和30b是交替剪接的,其编码蛋白VEGFR-3,该蛋白的胞外区域包括七个免疫球蛋白样重复域、一个跨膜区域及含有两个酪氨酸激酶结构域(TK1和TK2)的胞内段[14]。VEGFR3的胞外段与配体结合,活化的胞内段激活下游信号转导通路,从而调控淋巴管内皮细胞的增殖、迁移和生长。有研究总结既往报道PCL的38个已知FLT4基因变异位点,且增加20个新发变异位点,其中C.2797G>C和C.3122G>A已被报道5次,C.3323 _ 3325delTCT已被报道6次,其余大部分变异位点只被报道过1 次或2 次[5]。几乎所有报道的变异都位于该蛋白的TK区域(TK1和TK2,分别为17~20外显子和22~26外显子),而且大多数是错义变异,偶尔会有插入或缺失,从而导致酪氨酸激酶活性降低和淋巴发育受限[5]。然而,也有研究显示,具有典型临床症状及家族史的PCL患者中大约75%是由FLT4基因变异引起的,25%并无FLT4变异,故而认为可能存在其他尚未确定的遗传机制,也可导致与Milroy疾病表型一致的临床特征[7]。
另外,少数FLT 4基因变异位点位于编码TK 1、TK 2 以外的区域。最近报道1 例新的位于4 号外显子的剪接位点突变,c.361+5 G>A[15]。据推测,该变异会导致VEGRF3蛋白的分裂和提前截断,从而产生缺乏VEGRF同源域和c末端域的多肽,导致PCL的发生,这一研究在斑马鱼研究模型中得到了证实。近期报道一个新的位于17号外显子的错义变异,C.2515G>C,该位点编码氨基酸不在常规的TK 区域,而是位于胞质结构域,但推测因其与TK结构域较为接近,可能影响酪氨酸激酶活性,影响皮肤淋巴的发育[16]。本例患儿发现的FLT4基因变异c.3820 G>A(p.Asp1274Asn)为新发现的杂合变异位点,位于29号外显子,编码氨基酸位于TK 结构域以外的胞内区域,目前尚未见文献以及大规模人群频率数据库报道该位点变异。结合家系成员的临床表现及变异携带情况,推测该变异为致病变异。该变异对胞内段激活下游信号转导通路的影响机制有待进一步研究。
PCL 的诊断,除了需要与引起先天性胸腔积液、胎儿水肿的新生儿常见疾病相鉴别。还需要与其他引起淋巴回流受损而致淋巴水肿的各类遗传性疾病相鉴别,如MCLMR综合征(microcephaly with or without chorioretinopathy,lymphedema,or mental retardation)、特纳综合征、努南综合征、淋巴水肿-双行睫综合征、Meige病及黄指甲综合征等[17]。
目前,淋巴水肿尚缺乏有效的治疗方法。烘绑疗法、间歇加压疗法、手法淋巴引流等保守治疗对于预防淋巴水肿的形成及改善轻度淋巴水肿有一定的疗效。严重的水肿及后期皮肤纤维化等需行外科手术治疗。由于淋巴水肿的症状具有较大的个体差异,治疗需以早期长期综合个体化治疗为原则。对于PCL患者而言,预后相对较好,对于终身存在的淋巴水肿,适当适合的压缩袜、绷带、支持鞋、脚趾手套等是有益的,良好的皮肤护理是必不可少的。这些治疗措施可以改善肢体的美观,减小肢体的尺寸,降低并发症的风险。
综上所述,对于新生儿胸腔积液、胎儿水肿,在考虑常见疾病的基础上,有必要积极完善基因检测,以明确诊断、减少误诊及漏诊,便于及早干预、评估预后及指导遗传咨询。
猜你喜欢
中国现代医生(2022年21期)2022-08-22 03:30:42
世界最新医学信息文摘(2021年12期)2021-06-09 08:35:26
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:02
国际呼吸杂志(2019年4期)2019-03-12 01:08:06
中国医药科学(2017年9期)2017-08-04 21:42:14
放射学实践(2016年6期)2016-12-15 21:55:30
西南医科大学学报(2016年4期)2016-01-03 01:26:36
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:05
中国当代医药(2015年16期)2015-03-01 02:03:31
现代医药卫生(2014年18期)2014-03-11 19:33:24
