
吡喃酮类化合物的3D-QSAR及分子对接研究
2020-11-25 04:12:26吴鲁阳王天浩仝建波
陕西科技大学学报 2020年6期
吴鲁阳, 王天浩, 冯 怡, 张 星, 仝建波
(陕西科技大学 化学与化工学院 陕西省轻化工助剂重点实验室, 陕西 西安 710021)
0 引言
艾滋病,即获得性免疫缺陷综合症(Acquired Immune Deficiency Syndrome,AIDS)已经成为了当今时代仍未被攻克的医学难题之一[1].目前,在发展中国家,人类免疫缺陷病毒/获得性免疫缺陷综合症(HIV/AIDS)的流行对于公共卫生来说仍是一个巨大的挑战.根据联合国艾滋病规划署(UNAIDS)的报告,2017年全球有近3 690万人感染艾滋病病毒,94万人死于艾滋病,180万人新感染艾滋病病毒[2].我国卫生计生委发布的《2016年我国卫生和计划生育事业发展统计公报》指出,艾滋病依然占全国甲乙类传染病共报告死亡数的首位,占全部甲乙类传染病共报告死亡数的78.4%[3].
人类免疫缺陷病毒(Human Immunodeficiency Virus,HIV)是消灭艾滋病的主要障碍,如何有效地抑制该病毒是当前艾滋病的研究重点之一[4].人类免疫缺陷病毒可分为两种类型,即I型(HIV-1)和II型(HIV-2)[5-7],这两种类型的病毒之间存在一定的联系,而绝大多数AIDS都是由I型(HIV-1)病毒所引起的[8,9].目前,HIV的药物主要有逆转录酶抑制剂、整合酶抑制剂、蛋白酶抑制剂和进入抑制剂4种[10].
吡喃酮类化合物作为一类新型抗艾滋药物,具有良好的生物利用度,同时具备生产成本低、分子量小、结构简单等优点,因此具有良好的研究应用前景.如果能够进一步研究结构、优化设计分子并提高其生物活性,则有望成为一类新型高效抗艾滋药物[11].
三维定量构效关系[12]是一种应用广泛的设计药物分子的方法,它可以有效地把分子结构与其理化性质、结构参数、生物活性等结合起来.三维定量构效关系中具代表性的方法有CoMFA(Coparative molecular field analysis)与CoMSIA(Coparative molecular similarity index analysis)[13].
本文采用第二代CoMFA方法,即易位体比较分子场(Topomer CoMFA)[14]对18个吡喃酮类化合物进行3D-QSAR研究,建立了Topomer CoMFA模型,并通过Topomer Search技术[15]在ZINC数据库中进行R基团的虚拟筛选,设计出高活性的新分子,并采用分子对接[16]研究了新设计的分子与蛋白活性位点间的结合方式,在理论上为实际合成该系列新药物提供了理论参考.
1 实验部分
1.1 数据来源与分子结构
本文所研究的18个吡喃酮类化合物来源于文献[11,17].所有选取的18个化合物的分子结构骨架见图1所示,其结构和生物活性值见表1所示.采取隔三选一的方法将整个数据集分为训练集与测试集两部分,其中13个化合物作为训练集建立3D-QSAR模型,其余5个化合物作为测试集检验模型的预测能力.所有化合物的生物活性用pIC50(-logIC50)表示,IC50为抑制剂的半抑制浓度.

图1 化合物的结构骨架
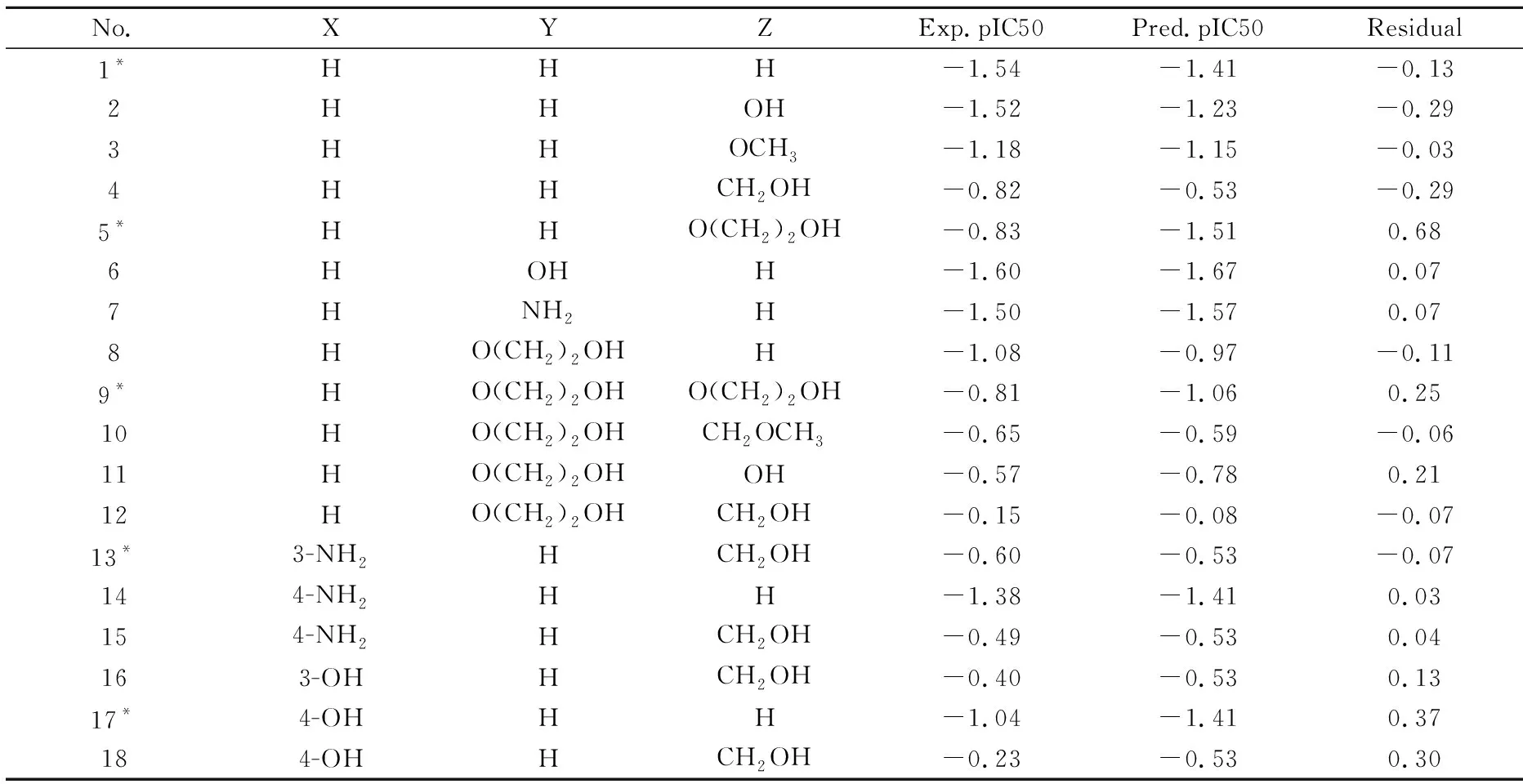
表1 化合物的结构和生物活性值
1.2 模型的建立
运用Powell能量梯度法,施加Tripos分子力场优化化合物分子[18],通过加载Gastelger-Huckel电荷,设定0.05 kcal/mol为能量收敛梯度值,1 000为最大迭代次数,剩余参数为SYBYL2.0-X默认值,取得分子的最低能量构象[19].
以训练集pIC50值最高的12号分子作为模板,通过两种方式对吡喃酮类化合物进行切割,并对其周围的立体场和静电场进行计算,利用偏最小二乘回归法(PLSR)对化合物进行建模.
1.3 虚拟筛选
本研究采用Topomer Search[20]技术,通过ZINC数据库中,筛选得到高活性贡献值的R基团.其中,Topomer CoMFA距离值为185.0,剩余参数为SYBYL2.0-X默认值.
筛选结果由Topomer CoMFA距离和R基团的活性贡献值体现,从数据库中筛选测出的R基团贡献值[21].一般在分子相似程度范围内,R基团贡献值越大,表明基团越有价值.
1.4 分子对接
采取Surflex-dock[22]技术进行分子对接,由PDB数据库提供对接所用的蛋白酶晶体结构,PDB数据库 ID为:1EE0,对接方式为SFXC.通过Total-Score、Crash和Polar函数评价分子对接结果[23]:Total-Score为总打分函数,分值越高表明受体与配体的亲和能力越好;Crash为碰撞打分函数,绝对值越趋近于零表明受体与配体对接的不适当程度越小;Polar为极性函数,结合位点在分子表面时,分值越高越好;结合位点在分子内部时,分值越低越好.
2 结果与讨论
2.1 建模的结果评价
通过Topomer CoMFA模型构建及预测化合物活性,以活性值最高的12号分子为如表2所示对化合物采用两种切割方式.
其中R1基团为蓝色,R2基团为红色,公共骨架为绿色.从表2可得出,两个模型的主成分数N分别为2和3,F(Fisher验证值)分别为19.799和23.879,r2(非交叉相关系数)均大于0.6,q2(交互验证系数)均大于0.5,SEE(标准估计误差) 分别为0.253和0.199,结果表明两种切割方式下构建的3D-QSAR模型都具有良好的拟合与预测能力,但模型2还较好的保存有化合物公共骨架,有利于R基团同公共骨架的选择.基于以上因素,选择模型2进行分子设计及对接研究.
图2为模型2下的18个化合物实验和预测活性值的线性回归图.图中各点均匀分布于45 °线两侧,模型2的预测和拟合能力较强.

表2 Topomer CoMFA建模结果
2.2 Topomer CoMFA三维等势图分析
三维等势图(图3)是在12号分子为模板的基础上,按模型2构建形成的.R1的立体场等势图和静电场等势图分别为图3(a)、3(b);R2立体场等势图和静电场等势图分别为图3(c)、(d).图3(a)、(c)绿色部分表示增大取代基的体积可提高化合物的活性,黄色部分表示减小取代基的体积可提高化合物的活性;图3(b)、(d)红色区域表明如引入带负电性取代基可提高化合物的活性,蓝色区域表明引入带正电性取代基可提高化合物的活性.
通过对图3(a)分析可知,R1取代基的位置上有大片绿色和小块黄色区域,表明绿色区域引入大基团取代基有利于活性的提高;表明黄色区域引入小基团的取代基有利于活性的提高.例如在黄色区域,化合物10(pIC50=-0.65)在R1取代基苯环的Z处用基团-CH2OCH3取代了化合物17、08、14、07、01、06(pIC50=-1.04,-1.08,-1.38,-1.50,-1.54,-1.60)上的-H、化合物04(pIC50=-0.82)上的-CH2OH以及化合物03(pIC50=-1.18)上的-OCH3,活性有显著提高.
由图3(c)可以看出,在R2取代基的位置上有大片绿色区域,说明在此处引入体积大的取代基有利于活性的提高,以及部分少许黄色区域,说明在此处引入体积小的基团有利于活性的提高.例如在黄色区域,化合物02(pIC50=-1.52)在R2取代基的位置上用-H取代了化合物06(pIC50=-1.60)的-OH取代基,活性相对提高;在大片绿色区域,化合物 08、10、11和12(pIC50=-1.08,-0.65,-0.57,-0.15)在R2取代基苯环的位置上用-OCH2CH2OH取代了化合物 03、14、02(pIC50=-1.18,-1.38,-1.52)上的-H、化合物07(pIC50=-1.50)上的-NH2和化合物06(pIC50=-1.60)上的-OH,活性明显提高.

(a)R1立体场等势图 (b)R1静电场等势图
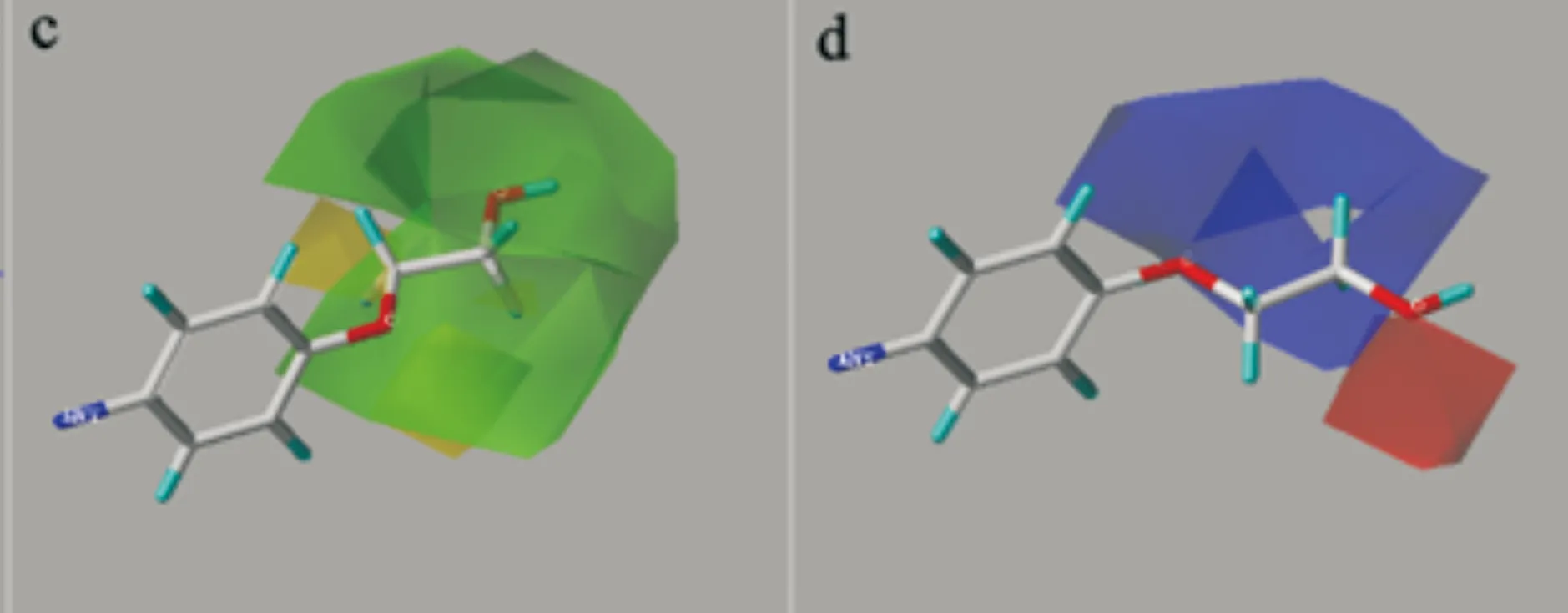
(c)R2立体场等势图 (d)R2静电场等势图图3 Topomer CoMFA三维等势图
2.3 分子设计
采用Topomer Search技术对R1和R2基团进行筛选,得到12个高活性的R2基团,同时选择12号分子的R1基团作为新分子的R1基团,共设计12个新的吡喃酮类化合物.通过Sketch Molecule绘制12个新化合物分子结构,并进行优化,化合物活性预测采用建立的Topomer CoMFA模型进行,新设计的12个化合物结构和预测活性值详见表3所示.
通过表3可知,01、02、03、04、05、06、12号共7个新设计的分子活性值高于模板分子.表明新设计的化合物理论上具有较好的抗艾滋病作用,可以作为抗艾滋药物的候选化合物.
2.4 分子对接
选择PDB数据库中与所研究的吡喃酮类化合物相关联的大分子蛋白1EE0作为对接构象,蛋白酶1EE0分类为转移酶(乙酰乙酰辅酶A络合的2-吡喃酮合酶).对1EE0进行预处理,提取出蛋白配体B/CAA700,通过创建形成活性口袋.如图4所示,表现出活性位点空间内配体小分子与大分子蛋白的相互作用.通过可行性验证分析,验证对接的准确性与可靠性.首先,导入提取的蛋白配体B/CAA700,对其加氢、优化;接着把配体作为分子直接对接到大分子蛋白1EE0上,对接结果如图5所示.得出配体B/CAA700构象(红色)与对接产生的1EE0对接构象(绿色)在空间内重叠程度良好,表明该方法准确可靠.
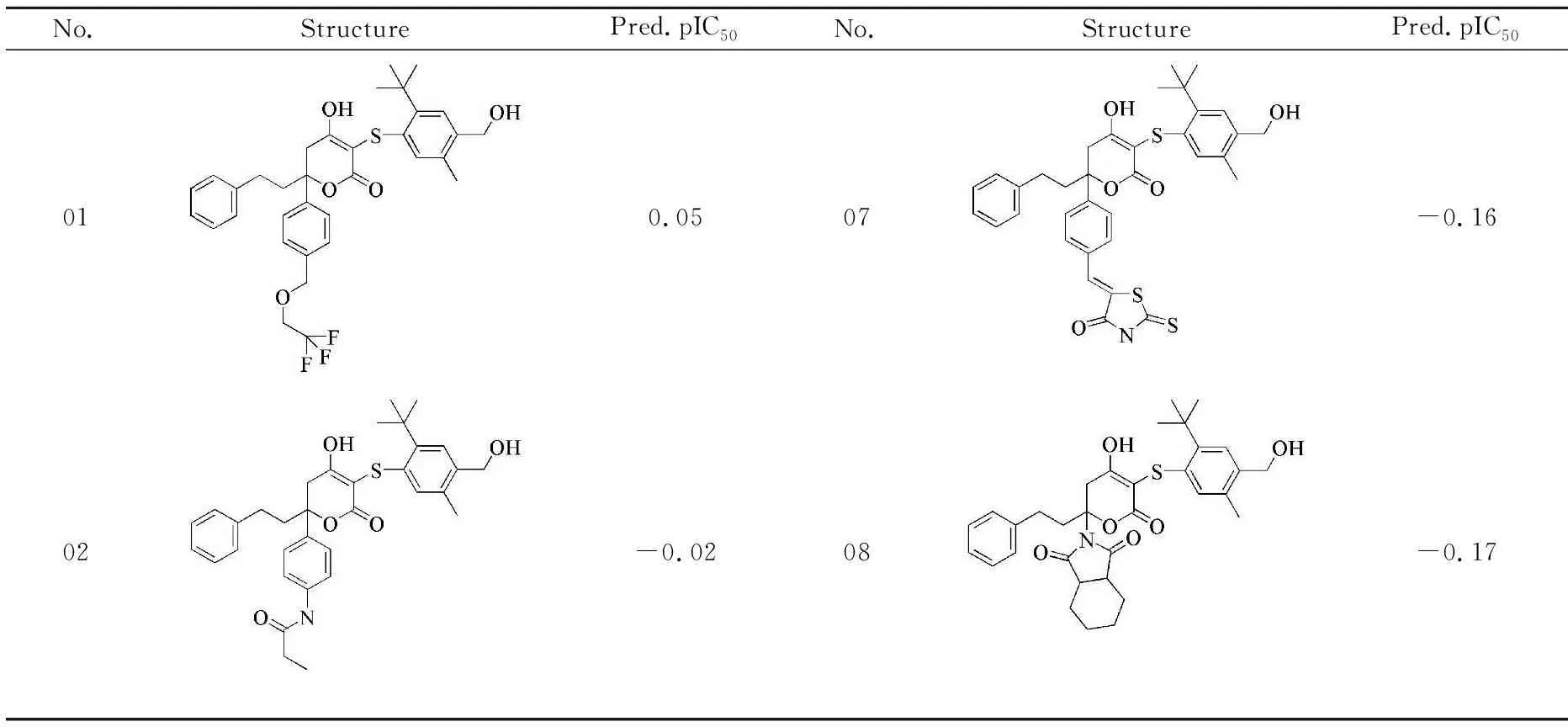
表3 设计化合物分子结构与活性预测值
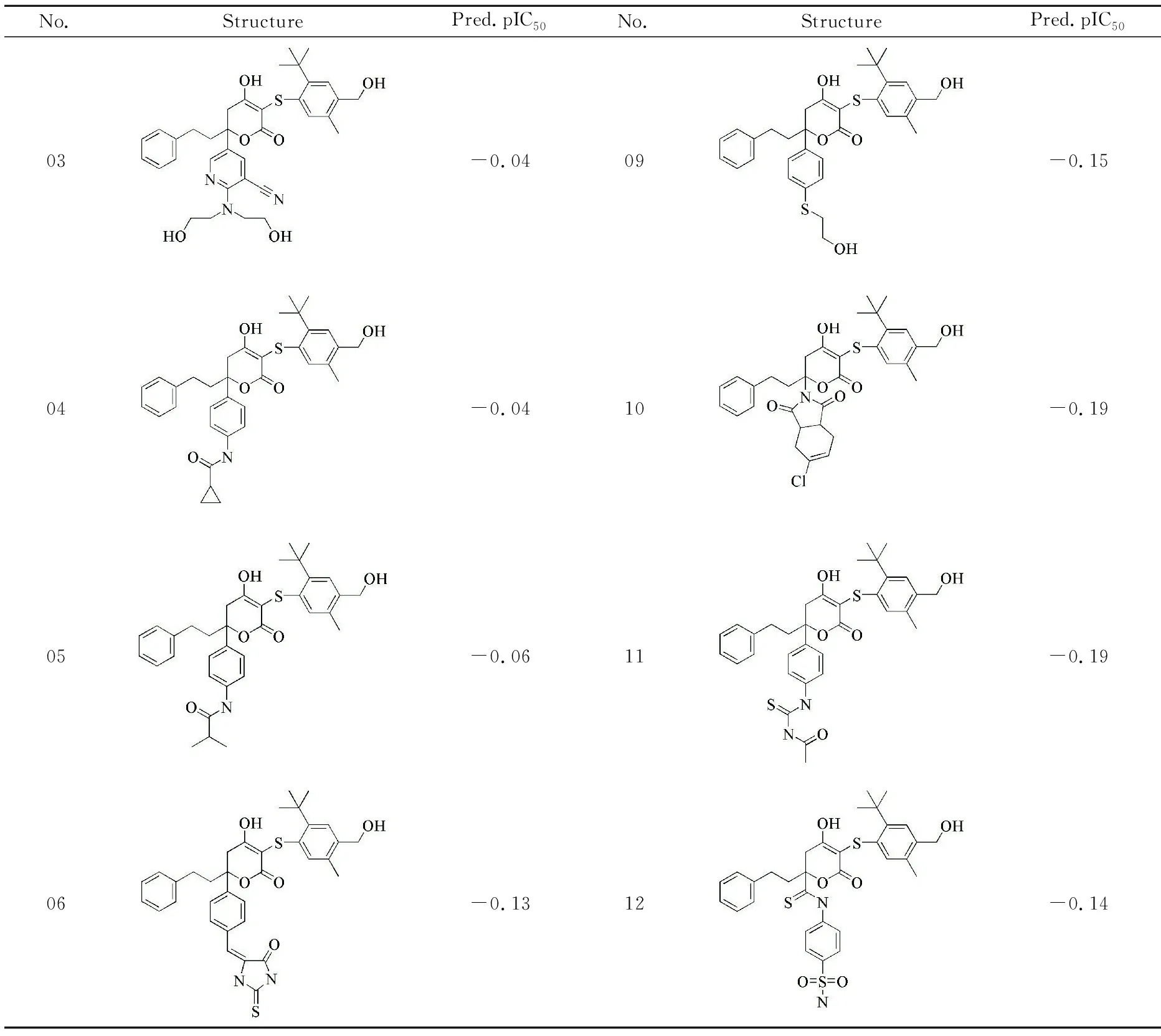
续表3
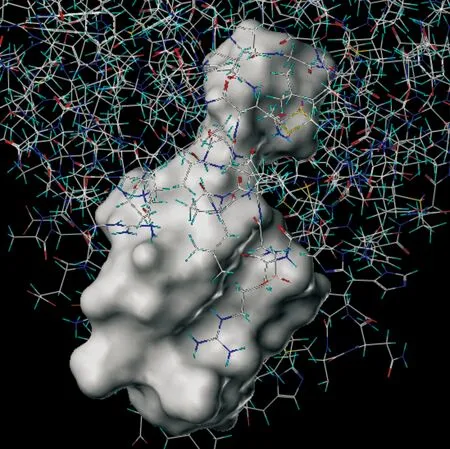
图4 原型分子生成图(灰色区域为原型分子)
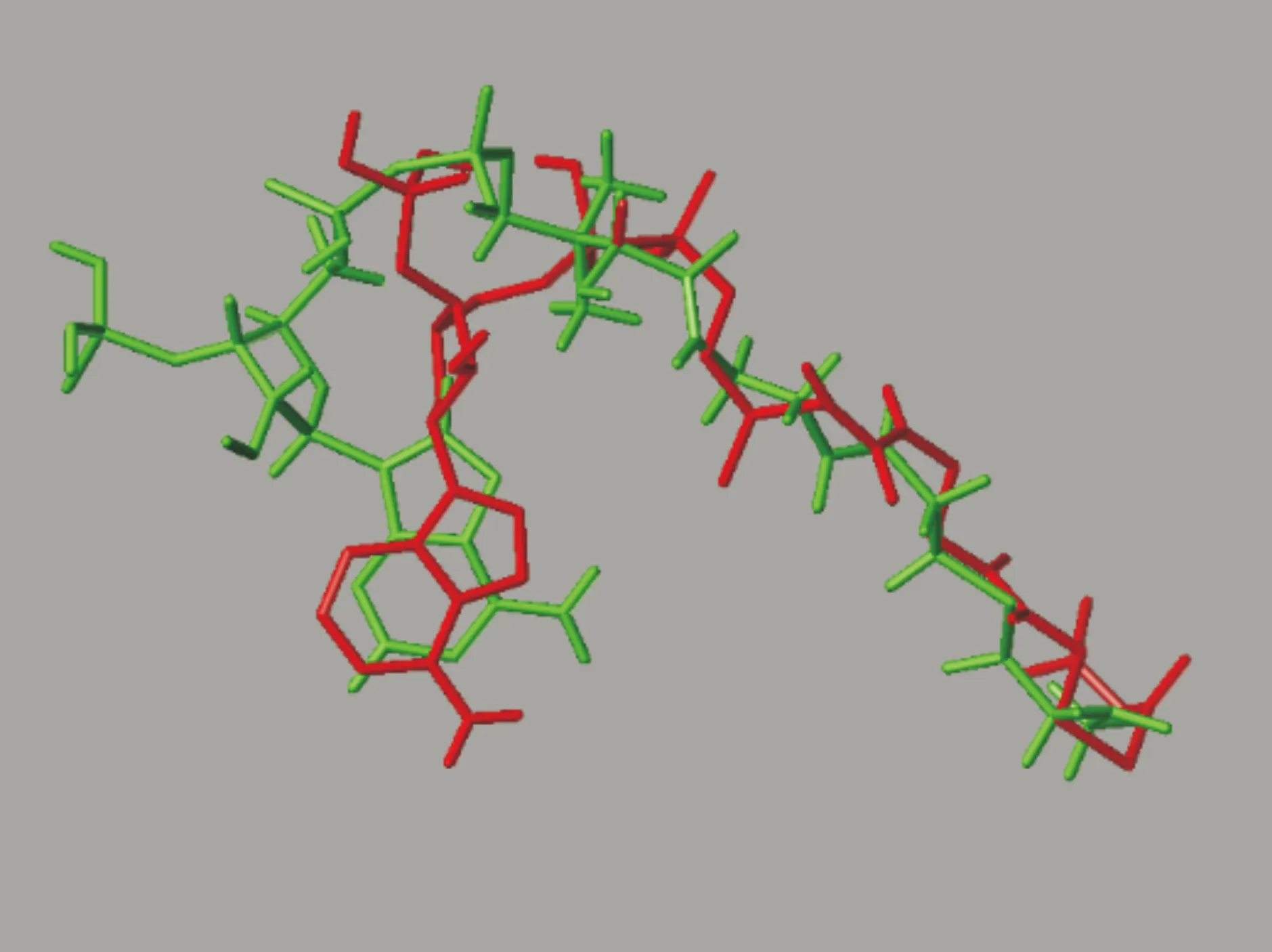
图5 晶体结构中配体构象与对接构象的对接模式
将活性值高于模板分子的7个新设计的分子与1EE0进行对接,共得到5个满足要求的对接模型.其中对接模型对应函数数值见表4所示.新设计分子01、03、04、05、06对接大分子蛋白活性位点的交互作用如图6所示.其中,绿色线状代表氨基酸残基,黄色虚线代表氢键.
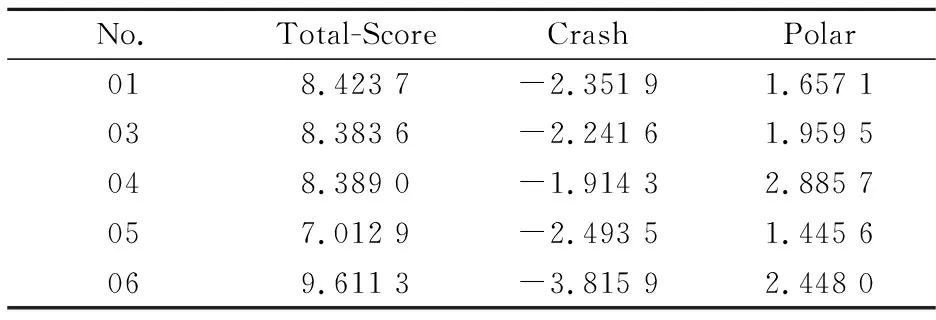
表4 分子对接结果
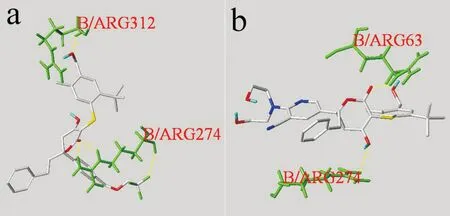
由图6(a)看出,对于01号分子,对接构象中O原子与B/ARG312中的H原子形成氢键,距离为1.927 Å;对接构象中F原子与B/ARG274中的H原子形成氢键,距离为2.459 Å;对接构象中O原子与B/ARG274中的H原子形成氢键,距离为1.926 Å,其形成另一个氢键距离为1.979 Å.
从图6(b)可知,对于03号分子,对接构象中O原子与B/ARG63中的H原子形成氢键,距离为1.852 Å;对接构象中O原子与B/ARG274中的H原子形成另一个氢键,距离为2.501 Å.
由图6(c)看出,对于04号分子,对接构象中O原子与B/ARG274中的H原子形成氢键,距离为2.040 Å,其形成另一个氢键距离为2.741 Å;对接构象中O原子与B/LYS67中的H原子形成氢键,距离为1.971 Å.
从图6(d)可知,对于05号分子,对接构象中O原子与B/ARG274中的H原子形成氢键,距离为1.978 Å;对接构象中O原子与B/LYS60中的H原子形成氢键,距离为2.153 Å,其形成另一个氢键距离为2.552 Å.
由图6(e)看出,对于06号分子,对接构象中O原子与B/ARG312中的H原子形成氢键,距离为1.548 Å;对接构象中H原子与B/THR68中的O原子形成氢键,距离为2.185 Å;对接构象中O原子与B/LYS67中的H原子形成氢键,距离为2.633 Å.
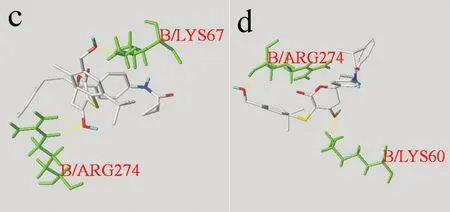
(a)、(b)、(c)、(d)、(e)分别表示新设计分子01、03、04、05、06号分子与1EE0的氢键作用图图6 氢键相互作用图
综上结果表明,一般配体分子与B/ARG274残基有氢键作用,如01、03、04、05号分子;配体分子与B/ARG312残基有氢键作用,如01、06号分子;配体分子与B/LYS67残基有氢键作用,如04、06号分子.
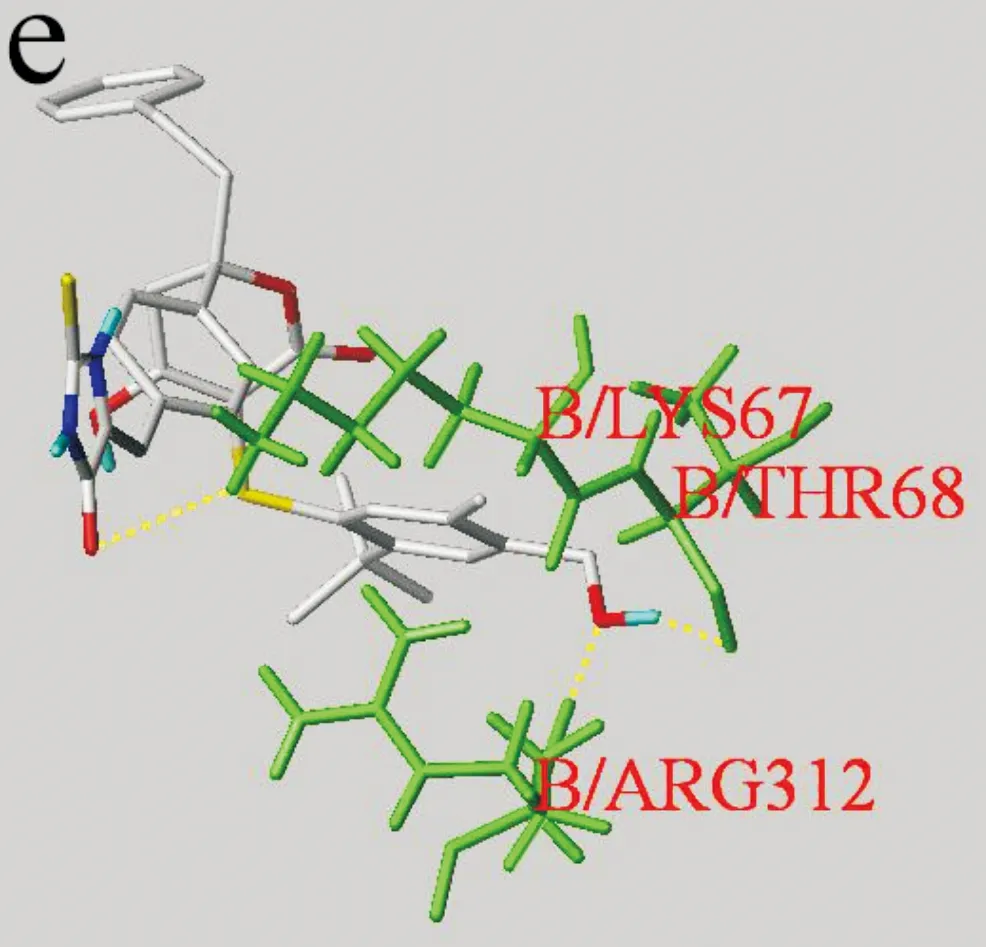
为了进一步说明这几种氨基酸残基的重要性,选择原数据集中活性最高的12号分子与活性最低的06号分子分别进行对接研究,其结果如图7所示.表明对接标准均符合要求.氨基酸残基B/ARG312、B/ARG274均与两个分子形成氢键;对于12号分子,还有氨基酸残基B/THR68、B/ARG63、B/LYS60与分子作用生成氢键,对于06号分子,还有氨基酸残基B/LYS67与分子作用生成氢键.

(a)、(b)分别表示12、06号分子与1EE0的氢键作用图图7 氢键相互作用图
3 结论
本文通过Topomer CoMFA 模型的建立,对18个吡喃酮类化合物进行了3D-QSAR研究,得到了良好稳定性及预测值的3D-QSAR模型.
首先,通过立体场与静电场,分析了不同取代基结构对活性的影响;然后,采用虚拟筛选技术从ZINC数据库筛选得到高活性基团,共设计出12个具备较高活性的新分子,其中7个分子活性高于原数据集分子,1个分子与模板分子活性相等;最后,利用Surflex-dock模块分析了5个分子配体及大分子蛋白1EE0之间的相互作用模式.
对接结果表明:配体与B/ARG274、B/ARG312、B/LYS67等氨基酸残基可以形成强的氢键相互作用,为抗艾滋病药物的进一步设计与合成提供了理论参考.
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
铜仁学院学报(2018年6期)2018-07-05 09:47:34
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
应用化工(2014年7期)2014-08-09 09:20:23
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年8期)2014-02-28 17:32:48
无机化学学报(2014年5期)2014-02-28 17:31:40
无机化学学报(2014年1期)2014-02-28 17:30:06
