
晚发型Pompe病患者的临床表现及基因突变特点
2020-11-23 03:59王敏卢岩邸丽徐敏陈海栾沁榕朵建英笪宇威
临床神经病学杂志 2020年5期
王敏,卢岩,邸丽,徐敏,陈海,栾沁榕,朵建英,笪宇威
Pompe病,即糖原累积症Ⅱ型(GSD Ⅱ),是一种罕见的常染色体隐性遗传的溶酶体贮积病,病因为编码α-葡萄糖苷酶(GAA)的基因突变,导致糖原在溶酶体内分解障碍,而不断蓄积在骨骼肌、心肌和平滑肌中。Pompe病根据发病年龄,受累器官,进展情况分为婴儿型(IOPD) 及晚发型(LOPD) 两种。LOPD患者的临床表现特征为肢体近端无力和呼吸衰竭[1]。
GAA基因位于染色体17q25.3,20 kb,包括20个外显子。所有GAA基因的变异和相关描述在Pompe病突变数据库中,含558个变异(www.pompecenter.nl)。一些变异可以在某些人群中多发,如c.-32-13T>G在高加索人群中突变比例可达34%~47%[2-3]。c.1935C>A(p.D645E)和c.2238G>C(p.W746C)在台湾人中多见[4],同时c.2238G>C(p.W746C)也常见于中国大陆LOPD中[5],c.1316T>A(p.M439K)和c.1857C>G(p. S619R)在韩国人LOPD中多见[6]。目前临床上对LOPD的认识不足,导致许多患者需要很长时间得到确诊。在此,本文对8例LOPD患者的临床表现、病理改变及分子遗传学基础进行了详细研究。
1 临床资料
1.1 一般资料 回顾分析2011—2019年收治的8例LOPD患者。其中,男5例,女3例;平均起病年龄16岁(9~21岁);平均确诊年龄24.8岁(14~46岁);病程2~30年。发病至确诊所需时间平均为8.9年。所有患者父母均非近亲结婚,2例有可疑家族史,其中1例患者同胞有突出的肢体无力症状(已死亡),1例母亲有类似病史。
1.2 临床表现 8例患者均以肌无力为主要表现,首发症状均为下肢无力,其中3例出现呼吸困难需要辅助呼吸,其他症状包括颈肌无力5例,躯干肌无力3例,所有患者可自主行走(表1)。
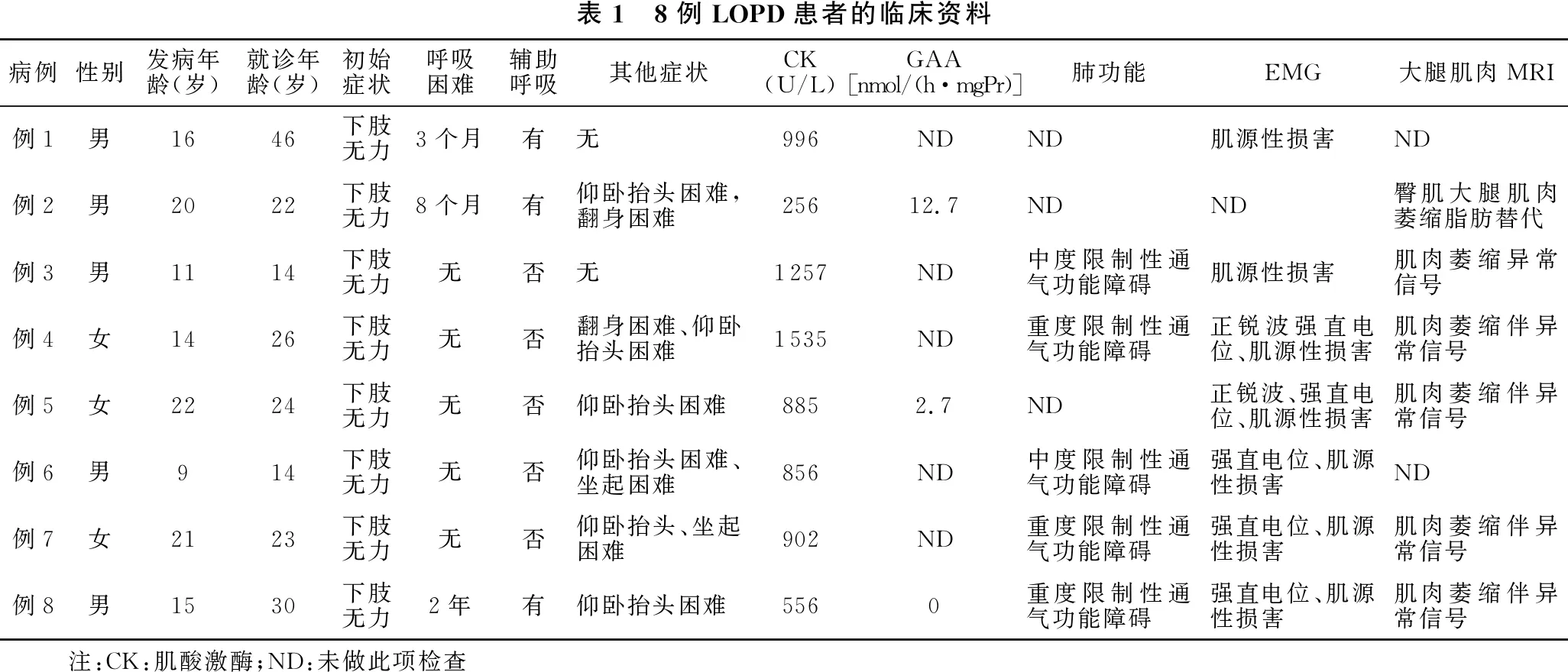
表1 8例LOPD患者的临床资料病例性别发病年龄(岁)就诊年龄(岁)初始症状呼吸困难辅助呼吸其他症状CK(U/L)GAA[nmol/(h·mgPr)]肺功能EMG大腿肌肉MRI例1男1646下肢无力3个月有无996NDND肌源性损害ND例2男2022下肢无力8个月有仰卧抬头困难,翻身困难25612.7NDND臀肌大腿肌肉萎缩脂肪替代例3男1114下肢无力无否无1257ND中度限制性通气功能障碍肌源性损害肌肉萎缩异常信号例4女1426下肢无力无否翻身困难、仰卧抬头困难1535ND重度限制性通气功能障碍正锐波强直电位、肌源性损害肌肉萎缩伴异常信号例5女2224下肢无力无否仰卧抬头困难8852.7ND正锐波、强直电位、肌源性损害肌肉萎缩伴异常信号例6男914下肢无力无否仰卧抬头困难、坐起困难856ND中度限制性通气功能障碍强直电位、肌源性损害ND例7女2123下肢无力无否仰卧抬头、坐起困难902ND重度限制性通气功能障碍强直电位、肌源性损害肌肉萎缩伴异常信号例8男1530下肢无力2年有仰卧抬头困难5560重度限制性通气功能障碍强直电位、肌源性损害肌肉萎缩伴异常信号 注:CK:肌酸激酶;ND:未做此项检查
1.3 实验室检查 所有患者均有肌酶升高,CK平均值905 U/L。3例患者进行了GAA酶活性检测,均明显降低,1例患者为0 nmol/(h·mgPr)。4例患者行基因检测,发现5种错义突变和2种无义突变,均为杂合突变,包括:c.-32-13T>G、c.1562A>T(p.E521V)、c.1634C>T(p.P545L)、c.1822C>T(p.R608*)、c.1839G>A(p.W613*)、c.1935C>A(p.D645E)、c.2238G>C(p.W746C)。
1.4 辅助检查 5例患者进行了肺功能检查,结果显示肺功能异常,呈中度或重度限制型通气障碍。7例患者EMG提示所检肌肉为肌源性损害,5例可见肌强直电位。所有患者行ECG及心脏彩超检查,均未发现心脏受累。6例LOPD患者行下肢的MRI检查,主要表现为大腿肌肉萎缩、受累肌群脂肪增生替代,部分肌肉可见水肿(图1)。8例患者均进行了肌肉病理检查,主要病理改变为部分肌纤维内出现较多空泡(图2),部分空泡内可见嗜碱性物质,PAS染色可见空泡内深染物质沉积,空泡肌纤维累及两型,以Ⅱ型肌纤维为主,部分肌纤维内出现NSE深染物质沉积。例4和例8患者肌肉病理除上述表现外,部分肌纤维内可见镶边空泡(图2)。

图1 例4(A)和例7(B)患者大腿MRI的T2WI-FS示不同程度的大腿肌肉萎缩,部分肌肉可见水肿,股二头肌短头、缝匠肌、股直肌、股薄肌(箭头)相对保存完好
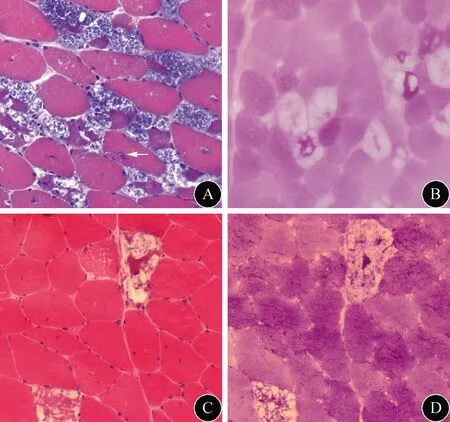
图2 例4(A)和例8(C)患者的股四头肌肌肉标本HE染色可见大空泡样变肌纤维,例4患者还可见镶边空泡(箭头所示);例3(B)和例8(D)患者PAS染色示部分空泡样变肌纤维糖原增多
1.5 治疗和预后 所有患者入院期间给予了对症治疗,患者病情无明显变化。
2 讨 论
Pompe病是一种罕见的代谢性肌病,呈常染色体隐性遗传或散发。GAA酶的缺失导致糖原在溶酶体内进行性累积、溶酶体膜破坏,糖原及破坏的水解物质进入细胞浆,替代肌肉的收缩物质,加之自噬作用的失衡,导致肌纤维破坏。病变主要累及心肌、骨骼肌及平滑肌。晚发型在1岁后出现症状,多表现为慢性进行性肢体近端无力和呼吸功能不全,心脏受累少见,呼吸功能衰竭是主要的致死原因。本文8例LOPD患者均于儿童期或成人期发病,首先表现为骨盆带肌和双下肢肌无力,爬楼梯或蹲起困难,有运动后疲劳现象,病情发展出现肩胛带肌和上肢无力,其中6例患者有仰卧抬头困难、3例有翻身坐起困难,中轴肌受累症状出现率达75%。提示除了肢体肌肉受累外,中轴肌受累也是LOPD患者临床特点,临床医生在采集病史和体格检查时应给予关注。本组病例就诊时需要间断辅助呼吸的LOPD患者有3例,其中1例患者多次发生上感引起的重症肺炎,甚至危及生命,值得重视。和本组其他患者一样,这3例患者也以下肢无力为首发症状,就诊时均可自行行走。进行肺功能检测的5例患者中,4例患者尽管病史中并无胸闷呼吸困难的主诉,肺功能检查却发现中度或重度限制性通气功能障碍,提示LOAD患者呼吸肌受累隐匿,并且可能发生在病程早期,容易被忽视。3例就诊时出现呼吸衰竭症状的患者,肢体运动功能相对保留,提示呼吸肌受累可以较四肢骨骼肌更严重,早期肺功能检测非常必要。
8例LOPD患者的平均起病年龄为16岁(9~21岁),平均确诊年龄24.8岁(14~46岁)。Byrne等[7]曾报道庞贝病登记处28个国家中的742例Pompe,LOPD患者的平均起病年龄为28.8岁,平均确诊年龄为37.1岁。而荷兰报道的54例患者中平均起病年龄和平均确诊年龄分别为28.1岁和35.4岁[8]。可以发现,本组病例的发病年龄较之明显提前,与其他来自中国大陆的报道情况相似。Liu等[9]曾报道5例中国大陆LOPD患者的平均发病年龄13岁,平均确诊年龄为22岁。刘祺等[10]报道6例LOPD患者,平均发病年龄8.4岁,确诊时平均年龄18.0岁。张寒冰等[11]报道的13例LOPD患者平均发病年龄9岁7个月,平均确诊年龄13岁8个月,提示中国LOPD的发病年龄普遍较高加索LOPD患者早。
肌肉MRI对于发现不同肌肉病肌肉受累和脂肪替代的特点非常有帮助。本组患者中6例LOPD患者的大腿MRI发现不同程度的大腿肌肉萎缩、受累肌群脂肪增生替代,部分肌肉可见水肿,表现为T2WI-FS上可见斑片状稍高信号。T2WI-FS上受累肌群主要包括前群的股外侧肌、股中间肌、股内侧肌,内侧肌群的大收肌,后组肌群的股二头肌长头,半膜肌受累较轻。以往有研究发现,Pompe患者早期大收肌、半膜肌多受累,半腱肌也可能受累,病情发展可能出现股二头肌长头、股内肌群的萎缩和脂肪替代,股二头肌短头大多不受累及,而缝匠肌、股直肌、股薄肌相对保存完好,甚至出现肥大[12]。本研究发现与之相符,但由于本研究的样本量有限,尚需要进行更大的样本研究。
本组8例患者的肌肉病理均具呈现Pompe典型病理特点,突出表现为部分肌纤维内边缘不规则的空泡结构,空泡大小及数量、形态各异,PAS 染色呈强阳性。值得注意的是,其中2例患者的肌肉病理除具有以上典型Pompe肌肉病理表现外,还有较多肌纤维内可见镶边空泡。Pompe病患者的肌肉病理出现镶边空泡最早被Schoser等[13]报道,在其报道的38例Pompe病患者中,镶边空泡为少见的病理表现。此后Dolfus等[14]报道了4例肌肉病理主要表现为镶边空泡的成人型LOPD患者,进一步酸性磷酸酶染色显示阳性,电镜检查发现充满糖原的溶酶体和自噬空泡从而确诊,提示镶边空泡可能是Pompe病的病理表现之一。对于不常见的病理表现,尤其以镶边空泡为主要表现的患者,要考虑有无LOPD可能。
GAA基因的突变位点多集中于三个区域:包含起始密码子的2号外显子;包含酶活性区域的10号和11号外显子;包含蛋白高度保守区域的14号外显子[15]。本研究发现的7个突变位点中只有两个位于11号外显子,其余位于1号内含子以及13号、14号和16号外显子,所有基因检测的患者均为杂合突变。-32-13T>G突变位点是高加索人中最常见的突变之一[3,16],突变位点c.2238G>C(p.W746C)位于16号外显子,为中国内地及中国台湾LOPD 患者的热点突变[5]。11号外显子的c.1562A>T(p.E521V)、c.1634C>T(p.P545L)以及14号外显子的c.1935C>A(p.D645E),已有报道见于中国Pompe患者[5]。
c.1822C>T(p.R608*)和c.1839G>A(p.W613*)均为无义突变,位于第13号外显子,导致蛋白翻译提前终止,对蛋白功能的影响可能较大。c.1839G>A(p.W613*)报道见于LOPD患者[17]。已报道的2例携带c.1822C>T突变位点的Pompe病患者,分别为美国人[18]和韩国人[6],此突变位点在中国大陆为首次报道。
本研究对8例LOPD患者的临床、病理表现和基因突变特点进行了分析总结,提示呼吸肌受累在LOPD患者中非常突出,要注意肺功能检测;对LOPD患者的不典型肌肉病理如镶边空泡要进一步检查。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
上海交通大学学报(2021年8期)2021-09-02
数字海洋与水下攻防(2021年2期)2021-05-08
国际放射医学核医学杂志(2021年10期)2021-02-28
食品安全导刊(2018年36期)2018-05-25
系统工程与电子技术(2016年2期)2016-04-16
汽车维护与修理(2016年3期)2016-02-28
船海工程(2015年4期)2016-01-05
能源(2015年8期)2015-05-26
