
慢性进行性眼外肌麻痹1例报告
2020-11-23 07:19:46路爱军董春霞赵倩倩曹秉振胡怀强
临床神经病学杂志 2020年5期
路爱军,董春霞,赵倩倩,曹秉振,胡怀强
慢性进行性眼外肌麻痹(CPEO)是一种以散发多见,临床主要表现为慢性进行性眼睑下垂、眼球活动障碍的线粒体疾病[1]。研究认为,mtDNA单一大片段缺失是散发性CPEO主要突变形式[2],相关病例报道较少。现将1例表现为肌肉线粒体基因单一大片段缺失所致的CPEO患者报道如下。
1 病例患者,男,22岁,学生,因“进行性吞咽呛咳15年,眼睑下垂、 复视伴四肢无力12年”于2018年2月入院。患者15年前无明显诱因出现逐渐加重的吞咽呛咳;12年前出现进行性加重的双眼上睑下垂,伴复视,快走或跑步时易疲劳,于当地某医院予以手术矫正,此后双眼睑下垂症状基本固定无改变,视物重影一直存在;5年前就诊于北京某医院,考虑动眼神经麻痹(具体治疗不详);4年前出现跑步不能,快走仅能坚持200 m左右;2年前出现言语笨拙,构音欠清,未予特殊治疗。发病过程中上述症状无晨轻暮重,无行走不稳、肢体抽搐、大小便障碍,无心慌、胸闷。既往体健,足月顺产,生长、智能发育正常。父母体健,否认有家族性遗传性和传染性疾病史。查体:体温36.2 ℃,心率72次/min,呼吸14次/min,BP 115/65 mmHg(1 mmHg=0.133 kPa),青年男性,发育正常,营养一般,体型消瘦,颈部细长,心肺腹部查体未见明显异常。神经系统查体:神志清楚,构音欠清晰,双眼内收、外展、上视、下视均受限,双侧瞳孔等大等圆,直径3.0 mm,直接和间接对光反射均灵敏,余颅神经检查无异常。抬头力弱,四肢肌力Ⅴ-级、肌张力正常,腱反射均未叩出,病理征阴性,指鼻试验及跟-膝-胫试验稳准,Romberg征阴性,深浅感觉无明显异常,脑膜刺激征阴性。实验室检查:肌酸激酶296.8 IU/L(24~194 IU/L)、乳酸5.50 mmol/L(0.7~2.0 mmol/L) 、同型半胱氨酸44.89 μmmol/L(0~15 μmmol/L)、叶酸2.73 μg/L(>3 μg/L)、维生素B12198 ng/L(180~914 ng/L);血常规、肝功能、血脂、生化、甲状腺功能、乙酰胆碱受体抗体、乙酰胆碱酯酶抗体均正常。新斯的明试验阴性。ECG示窦性心动过缓;心彩超示心内结构未见明显异常;胸部CT示甲状腺、胸腺未见异常;EMG示所检肌肉呈肌源性损害,重复神经电刺激未见异常改变;头颅MRI平扫未见明显异常。经患者同意于局麻下行肱二头肌活检,HE染色可见多个肌束,肌纤维大小不等,散在小圆纤维,可见较多肌膜下呈紫红色肌纤维,其胞浆内有不规则裂隙或小空泡(图1A);MGT染色可见较多散在分布的破碎红纤维(RRF)(图1B);NADH示两型肌纤维分布大致正常,部分肌纤维肌膜下NADH酶活性增高;COX染色见部分肌纤维COX酶缺失(图1C、图1D);ORO、PAS染色未见特征性改变。当时经患者同意,采血行明睿_focus_线粒体基因组检测,检测了线粒体全基因的16 569个位点,在已报道明确致病的66种突变中未发现异常。2019年10月取患者的肌肉行金准_focus_线粒体基因组检测,检测了该样本线粒体全基因的16 569个位点,在已报道明确致病的89种突变中未发现异常;但该样本在线粒体基因组发现大片段缺失突变,缺失区域为chrM:8482-13446(图2)。因患者家庭原因,没有采集到患者父母的血液标本,未能行基因的家系验证。

图1 肱二头肌活检。HE及MGT染色可见散在的破碎红肌纤维,COX染色见部分肌纤维COX酶缺失。A:HE染色(×400);B:MGT染色(×400);C:COX染色(×100);D:COX染色(×400)
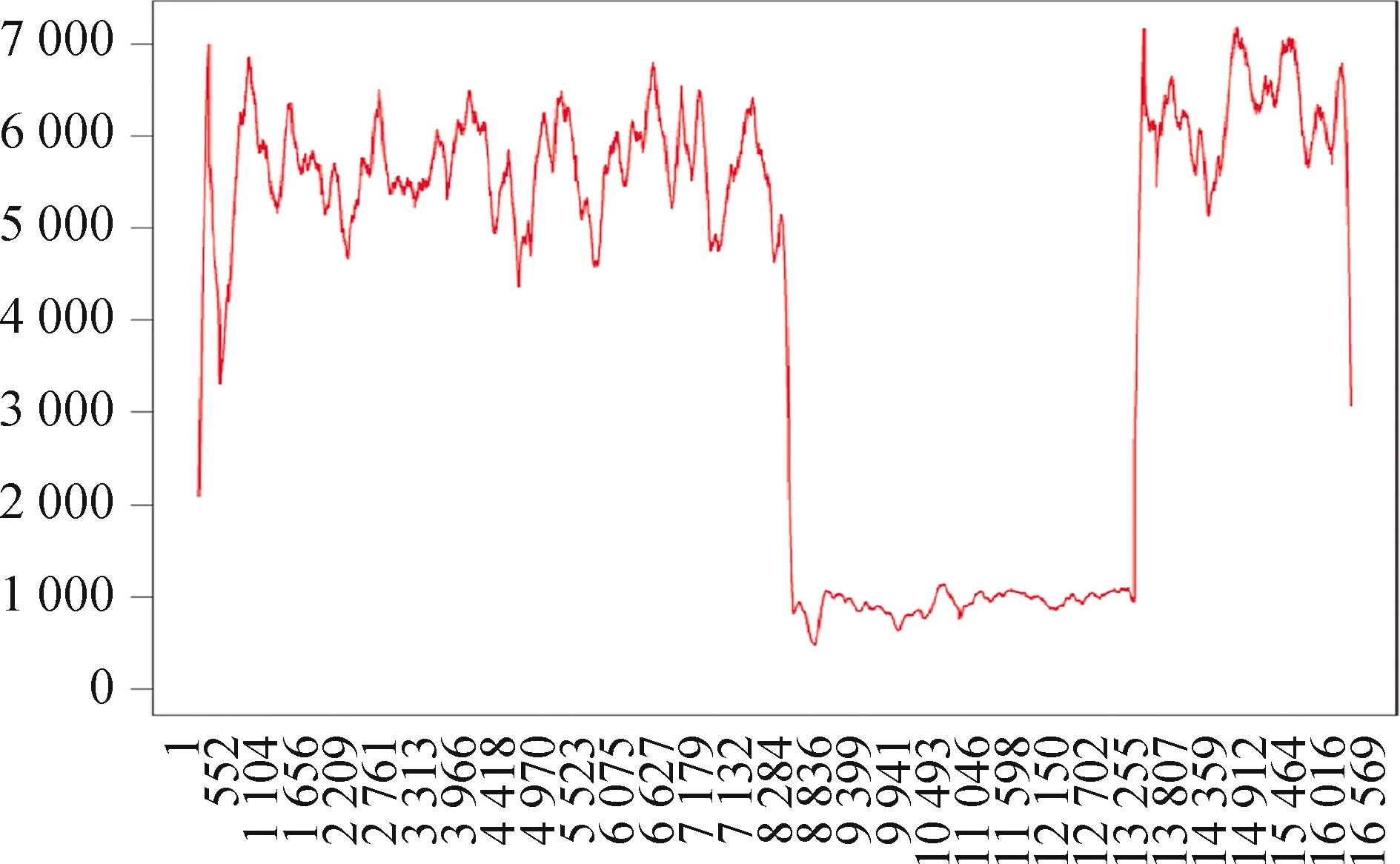
图2 骨骼肌线粒体基因检测结果。患者存在chrM:8482-13446大片段缺失突变
2 讨论CPEO可见于任何年龄,30岁前起病多见[3],多呈散发性,常以上睑下垂首发,个别患者首发症状为复视、不耐受疲劳,临床主要表现为进行性上眼睑下垂、眼球运动障碍、不耐受疲劳和肢体近端无力。因双侧眼外肌基本对称性麻痹,而且病情进展缓慢,各眼外肌多出现均衡代偿,大多数患者无复视症状[4]。构音障碍在CPEO报道中少见。本例患者儿童期起病,以吞咽障碍首发,继而出现眼睑下垂、复视,伴有不耐受疲劳,并呈进行性加重,后出现构音障碍。研究表明,mtDNA单一大片段缺失是散发性CPEO主要突变形式,但缺失片段长度与临床表型无显著相关性[2], mtDNA点突变或nDNA突变也可引起该病[5]。本例患者EDTA全血基因检测mtDNA未发现异常。mtDNA具有异质性和有丝分离性,即野生型和突变型mtDNA可存在于同一类细胞中,但随细胞分裂而分配到不同细胞,最终可能达到同质性,分裂旺盛的细胞如血细胞往往有排斥突变型mtDNA的趋势[6],最终导致在外周血细胞中只有野生型或仅有少量的突变型mtDNA。Schon等[7]研究发现,半数CPEO患者的骨骼肌细胞中有mtDNA基因缺失,缺失片段在1.3~7.6 kb之间,其中三分之一的患者有5 kb的缺失,缺失的起点是第8 483位碱基,终点在13 459位,共有4 977 bp,称之为“共有缺失”。为此,又对患者的骨骼肌进行了线粒体基因检测,结果发现患者骨骼肌线粒体基因组存在大片缺失突变,缺失区域为chrM:8482-13446,与文献报道的较一致[7]。
骨骼肌活检是诊断线粒体病的重要方法。RRF是由线粒体结构和功能障碍后大量线粒体反应性增生聚集所致,是线粒体疾病最典型的病理表现[8]。在儿童患者中COX阴性纤维可能比RRF更多,或者可能是肌肉活检中唯一的异常[9]。由于CPEO伴肌无力患者四肢和躯干肌肉受累相对较轻,肌活检有时不能发现RRF,这种情况下发现COX缺失纤维对CPEO的病理诊断具有重要价值。该患者肌肉活检病理既有RRF,又有大量COX缺失的肌纤维,纵断面可见在同一个肌纤维内COX成灶性缺失,组织病理符合CPEO的病理特点。
CPEO的早期诊断误诊率较高,诊断过程中一定要注意与临床常表现为眼外肌麻痹的重症肌无力、Kearns-Sayre综合征、眼咽肌型营养不良等疾病鉴别。重症肌无力可伴有复视,呈晨轻暮重,新斯的明试验阳性,血清中乙酰胆碱受体抗体明显升高,重复神经电刺激动作电位波幅递减,且低频刺激递减程度在10%以上、高频刺激递减程度在30%以上,胸腺CT可见胸腺增生、肥大或肿瘤。KSS是线粒体病的一种亚型,多在20 岁以前发病,进行性眼外肌麻痹,视网膜色素变性,常伴有心脏传导阻滞、小脑性共济失调,还可有身材矮小、神经性耳聋、智力减退等[10]。眼咽肌型营养不良呈常染色体显性遗传,多在40岁以后起病,咽喉肌症状突出,伴眼外肌麻痹,肌活检、基因检测可以与线粒体病相鉴别。因此,临床怀疑本病时应尽早行肌肉活检,对其早期诊断具有重要意义。分子生物学是线粒体病诊断的重要手段,基因检测是其诊断的金标准[11]。在标本的选择中肌肉组织优于血液,单纯的血液基因检测未发现异常并不能除外该疾病。基于临床表现及骨骼肌病理检查,选择合适的标本组织,有助于线粒体病相关nDNA和mtDNA的基因检测,可从不同水平对线粒体病予以诊断。目前国内对mtDNA单一大片段缺失所致线粒体病患者的报道较少,尚缺乏系统性的临床、病理及分子遗传学研究。
猜你喜欢
中国医药指南(2024年2期)2024-01-25 16:53:54
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
国际眼科杂志(2021年4期)2021-04-12 07:14:16
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
甘肃科技(2020年20期)2020-04-13 00:30:54
中国心血管杂志(2020年4期)2020-01-09 03:47:19
医学信息(2018年5期)2018-04-20 11:03:20
天津农学院学报(2016年2期)2016-12-01 05:40:05
中国当代医药(2015年26期)2015-03-01 02:07:07
