
新生儿点状软骨发育不良1例并文献复习
2020-11-18 08:02:48段佳佳王举李书津郭菲菲徐发林
河南医学研究 2020年30期
段佳佳,王举,李书津,郭菲菲,徐发林
(郑州大学第三附属医院 新生儿科,河南 郑州 450002)
新生儿点状软骨发育不良又称为先天性钙化性软骨发育不良,是以骨骺软骨的不规则钙盐沉积为病理特征的一类骨发育异常性疾病。主要临床表现为身材矮小、面部畸形、四肢短缩,大部分伴有生长发育迟缓、智力低下,部分患者可有白内障、鱼鳞病等[1-2]。本病发病率低,临床罕见,国内外报道极少,因此临床医生往往对本病认识不足,易导致误诊或漏诊。患儿多因呼吸道症状就诊,面部发育异常及全身骨骼多发性斑片状钙化影是提示本病的首要诊断线索。详细询问病史及体格检查发现身材矮小、四肢短粗、心脏杂音、听力异常及其他表现等可做出临床诊断。本文对郑州大学第三附属医院2017年8月收治的1例新生儿软骨发育不良患儿的临床特点进行分析,并复习相关文献,希望对提高本病的认识有所帮助。
1 病例资料
患儿,男,出生21 h,以“鼻塞21 h,发现呼吸困难4 h”为主诉入院,系第2胎第2产,36+5周,因“胎心慢”行剖宫产,出生时体质量3 300 g,羊水多,胎盘、脐带正常。1、5分钟Apgar评分均为10分。出生后即发现患儿鼻骨塌陷,出现鼻塞症状,给予保暖等对症处理后鼻塞无明显缓解,出生后17 h发现患儿出现呼吸急促,呼吸困难进行性加重转入郑州大学第三附属医院。入院查体:反应欠佳,前囟平软,头围33 cm,鼻梁塌陷,眼距稍增宽,扁平脸,耳位较低,颈短(图1),高腭弓,胸廓稍饱满,呼吸急促,吸气三凹征阳性,双肺呼吸音粗,未闻及湿啰音。心率每分钟142次,律齐,胸骨左缘2~3肋间可闻及Ⅱ级收缩期杂音。身长43 cm,四肢短粗(图2),余查体未见明显异常。其一姐姐3岁,体健。母亲3 a前确诊为系统性红斑狼疮,规范治疗至今。孕期四维超声(孕24周)示:鼻骨低平,额鼻夹角增大,两鼻孔间距增宽,上牙槽宽平;染色体微阵列未见明显异常。胎儿心动超声(孕30周时)示:动脉导管流速增快,右心室稍增大,心包积液。
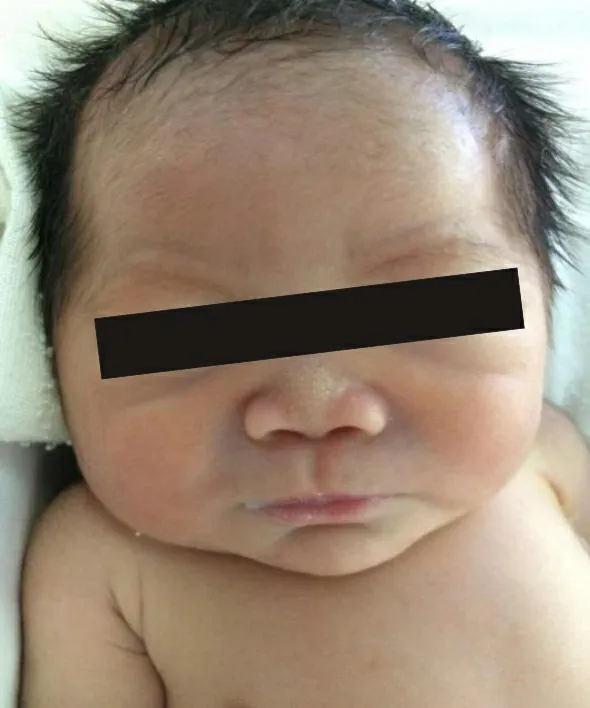
照片已征得患儿家属同意。
入院后检查:血气分析pH 7.41,PO253 mmHg(1 mmHg=0.133 kPa),PCO236 mmHg BE -2.2,SPO291%。胸片示:早产婴肺,左侧胸廓塌陷,第5、6肋骨形态欠佳,所见椎体及附件形态失常,见多发斑点状高密度影(图3),入院后给予经鼻特续气道正压通气氧疗,肺表面活性物质360 mg气管导管内注入,患儿呼吸状况逐渐好转,3 d后改为鼻导管,后停氧。辅助检查:全脊椎正侧位片示:脊柱生理弯曲存在,未见明显侧弯,诸椎体、椎弓根形态异常、骨质密度不均,呈斑点状骨质沉积,椎弓根间距变窄;侧位片显示椎板形态及密度尚可,双侧肋骨计数12对,肋骨骨密度及形态尚可,提示:椎体软骨性成骨障碍,考虑遗传代谢性骨病(图4)。四肢长骨平片示:四肢长骨骨皮质变薄,牙腔密度稍增高,左侧股骨中分局部骨皮质连续性欠佳,双足诸骨密度减低,呈多发颗粒样骨质改变,提示四肢及双足诸骨质结构改变,首先考虑系统性骨病,左股骨骨折待排(图5)。心脏超声示:房间隔缺损(2.5 mm),动脉导管未闭。肝胆胰脾及泌尿系超声未见异常。耳声发射双耳均通过,眼科会诊未见明显异常。染色体核型分析未见异常。基因检测提示点状软骨发育不良,考虑CDPX1型。
入院后根据患儿X线检查及特殊面容,诊断为:(1)新生儿点状软骨发育不良(CDPX1型);(2)新生儿呼吸窘迫综合征;(3)早产儿。患儿治疗1周后停氧可耐受,奶量每次25 mL,每2 h可按时完成,与患儿家属沟通后家属放弃治疗,要求出院。出院后随访至今,患儿生长发育迟缓,易患呼吸道感染。

图3 患儿脊柱正位片 图4 患儿脊柱侧位片

图5 患儿四肢长骨正位片
2 讨论
点状软骨发育不良首次报道见于1914年,临床较为罕见[3]。本病根据遗传方式及临床表现可分为5型:常染色体显性遗传型(非肢根型或conradi-hunermann病)、常染色体隐性遗传型(肢根型,rhizomelic chondrodysplasia punctata,RCDP)、X连锁隐性遗传型(CDPX1型)、X连锁显性遗传型(CDPX2型或conradi-hunermann-Happle型)以及Sheffield型。各型发病机制及临床表现差异较大,目前尚不十分清楚,其主要鉴别依赖于基因检测。非肢根型目前发病机制尚不清楚,遗传方式为常染色体显性遗传,多无皮肤病变及眼部异常[2]。肢根型是目前了解最多的类型,主要是由于PEX7基因突变所致的过氧化物酶体代谢缺陷,导致过氧化物酶体靶序列2介导的过氧化物酶体基质转运异常,尽管该类型患儿的过氧化物酶体中存在过氧氢酶,但其过氧化物酶体结构存在异常[4]。该型主要临床表现为外观异常、身材矮小、生长迟缓、智力障碍,常合并眼部病变和先天性心脏病等,是最严重的一种类型。CDPX1系X连锁隐形遗传,主要是位于Xp22.3上编码芳香基硫酸酯酶E的基因突变导致 ARSE 活性改变所引起[5]。该型患儿常有鼻腔发育畸形,特殊面容,呼吸困难,远端指指短缩,可合并气管支气管狭窄,合并先天性心脏病及眼部疾病较少[6]。CDPX1也可能与维生素K依赖的芳基硫酸酯酶E缺陷引起[7],此型易与Keute综合征混淆,也可见于患有自身免疫性疾病母亲的患儿[8-10]。CDPX2型为X连锁显性遗传,是由于EBP基因缺失突变导致,临床表现为非对称性肢体缩短,伴有皮肤损害,常在婴儿期死亡[11]。Sheffied型目前研究较少,其遗传方式及发病机制尚不明确,需进一步研究。
点状软骨发育不良的临床表现主要包括[1,4]:(1)特殊面容,扁平脸、眼距增宽、鼻梁塌陷、腭裂、短颈;(2)四肢畸形,短肢、多指、并指、身材矮小、四肢短粗、关节挛缩或脱位;(3)皮肤病变,毛发脱落、鱼鳞状角化症;(4)眼部异常,斜视,眼球震颤、白内障、视神经萎缩或发育不良。典型X线表现为在长骨、椎骨、肩胛骨处出现骨化异常,骨质密度不均,骨骺端斑点状钙化等,部分患儿关节软组织处也可见斑点状钙化影。同时该病可能合并各种先天性心脏病,如室间隔缺损、房间隔缺损、动脉导管未闭或其他先天畸形。部分患儿可合并不同程度的听力损伤。本例患儿孕期四维超声(孕24周)即发现胎儿鼻骨低平,额鼻夹角增大,两鼻孔间距增宽,上牙槽宽平。出生后体格检查发现患儿面容特殊,鼻梁塌陷,眼距增宽,扁平脸,耳位较低,颈短,高腭弓,胸廓稍饱满,四肢短粗,呼吸急促,吸气三凹征阳性,胸骨左缘2~3肋间可闻及Ⅱ级收缩期杂音。心脏超声提示该患儿存在先天性心脏病(房间隔缺损、动脉导管未闭)。同时X线检查发现该患儿脊椎及四肢长骨骨化不全,可见多发斑点状钙化影,这些体格检查及辅助检查符合该病的特征性表现。但本例患儿并未发现存在严重的听力损伤及眼底病变,远期仍需进一步随访。
本例患儿全身X线检查发现患儿脊椎及四肢长骨骨化不全,可见多发斑点状钙化影,结合患儿身材矮小、特殊面容,孕期染色体微阵列检查未见明显异常,生后染色体核型分析未见异常,行基因检测提示点状软骨发育不良,考虑CDPX1型。患儿治疗1周后停氧可耐受,呼吸平稳,奶量每次25 mL,每2 h 1次可按时完成,患儿病情好转后出院。本例患儿母亲系系统性红斑狼疮患者,长期口服小剂量激素规范治疗,通常在孕期选用泼尼松甲强龙来治疗,该药物可被胎盘代谢,且在母体药物剂量小于20 mg时,仅10%进入胎儿循环,而长期应用糖皮质激素会导致骨发生及骨丢失,从而影响骨的形成。故该例患儿的发病是否与其母亲系系统性红斑狼疮患者,长期服用糖皮质激素有关,从而影响了骨的形成,查阅多方面相关资料后发现系统性红斑狼疮合并妊娠主要是增加了母亲患其他疾病的风险,如高血压、糖尿病、子痫等,对胎儿的主要影响是流产、死产、新生儿死亡、早产和胎儿宫内生长受限等不良事件[12-14],而类似于该患儿(全身多部位的点状软骨发育不良)的疾病表现国外鲜有报道,目前国内尚未见相关文献报道。
点状软骨发育不良会累及多脏器损害,目前无特效治疗方法,以对症治疗为主。根据不同临床特点,建议行基因检测以明确分型,不同分型,预后不同。各型差异较大,大多数患儿可以长期存活,但成年后往往身材矮小。其中肢根型预后最差,常伴有严重的智力低下,多数此型患儿最终因呼吸衰竭于婴儿期死亡。
由于新生儿点状软骨发育不良临床罕见,因对本病缺乏认识,往往导致误诊或漏诊。患儿多因呼吸道症状就诊,面部发育异常及全身骨骼多发性斑片状钙化影是提示本病的首要诊断线索。详细询问病史及体格检查发现身材矮小、四肢短粗、心脏杂音、听力异常及其他表现等可做出临床诊断。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31 09:57:56
建材发展导向(2022年14期)2022-08-19 02:08:40
今日农业(2022年1期)2022-06-01 06:18:04
小雪花·初中高分作文(2019年8期)2019-10-07 08:46:42
新医学(2019年4期)2019-09-10 07:22:44
四川冶金(2017年2期)2017-04-11 12:55:36
中国卫生标准管理(2015年16期)2016-01-20 09:26:16
哈尔滨医药(2015年5期)2015-12-01 03:58:08
安徽医科大学学报(2015年11期)2015-07-20 02:41:08
中国医药科学(2015年6期)2015-07-18 01:30:09
