
极端环境用中温固化改性氰酸酯树脂制备及性能
2020-11-14 01:06吴鑫锐高堂铃王荣国吴健伟曹殿学
固体火箭技术 2020年5期
王 冠,吴鑫锐,高堂铃,邵 南,付 刚,王荣国,张 斌, 吴健伟,匡 弘,曹殿学
(1.黑龙江省科学院 石油化学研究院,哈尔滨 150040;2.哈尔滨工程大学 材料科学与化学工程学院,哈尔滨 150001;3.上海复合材料科技有限公司,上海 201112;4.哈尔滨工业大学 复合材料与结构研究所,哈尔滨 150080)
0 引言
未来新一代先进航天飞行器运行环境更加苛刻(-200~200 ℃,空间辐照等),这就要求某些特殊结构的复合材料需在较宽的温差范围内具有尺寸和强度的稳定性并且阻燃,而现今航天工程中大量应用的环氧树脂基复合材料,因其树脂基体的耐温性(Tg<200 ℃)、阻燃性较差,已不适合于先进航天器某些特殊结构的复合材料设计中[1-3]。近年来,氰酸酯树脂(CE)由于固化后形成独特的三嗪环结构且交联点间通过醚键相连,具有优异的耐高温稳定性(Tg>300 ℃)、良好的高、低力学强度、工艺性能和较好的阻燃性,具备了可在深空环境稳定应用的潜质[4-5]。由于氰酸酯树脂交联密度较大,单独使用时其固化物的韧性较差,无法满足极端环境下的应用。另外,单纯氰酸酯树脂的固化温度较高,一般需要经过200 ℃以上的后处理才能获得较好的耐温效果,但是过高的加工温度导致制备大尺寸复合材料构件过程中残留较多的内应力,从而降低制件的尺寸精度,影响了复合材料制件在极端环境下应用的稳定性。因此,为提升氰酸酯树脂在空间极端环境用复合材料中的应用潜力,需要对氰酸酯树脂进行改性提高其低温韧性和降低其固化成型温度,实现中温固化(120~140 ℃)。
现阶段增韧改性氰酸酯的方法通常是在氰酸酯网络中引入液体橡胶或热塑性工程塑料,利用固化微分相技术可大幅度地提高氰酸酯树脂的韧性,其中室温断裂韧性可提高2.5~3.2倍[6];但是,通过上述方法改性得到的氰酸酯固化物经室温至超低温(-150 ℃以下)冷热循环过程时,在两相界面产生内应力,最后过多内应力积累超过树脂本身的强度导致树脂基体的破坏,从而达不到低温增韧效果[7]。黑石化的王冠等[8],曾采用聚酚氧树脂和环氧树脂共改性氰酸酯树脂,改性后氰酸酯树脂基体在-196 ℃下冲击韧性可达18.8 kJ/m2,玻璃化温度超过210 ℃,经过15次冷热循环(-196~200 ℃)未见裂纹出现,获得较好低温力学性能,但是,此树脂体系采用高温(200 ℃)固化。有文献报道[9],采用有过度金属盐、有机锡和活泼氢催化等能大幅降低氰酸酯树脂的固化温度,实现氰酸树脂的中温固化,但是采用上述方法得到氰酸树脂室温贮存期差(不超过7 d),无法工程化制造。郑亚萍等[10]采用脲类衍生物有效降低环氧树脂的固化温度,实现中温固化且有适中的室温贮存期。国内外文献对于脲类衍生物用于氰酸酯树脂体系降低其固化温度的研究尚未见报道。
结合前期对于氰酸酯树脂在极端低温环境下应用的研究成果,本文采用纳米配体与单异氰酸酯制备负载脲类衍生物促进剂(M),通过热熔共混的方式加入到聚酚氧/环氧树脂共改性氰酸酯树脂体系中,制备了极端环境用中温固化改性氰酸酯树脂,研究了改性后氰酸酯树脂的固化行为和树脂体系流变行为。考察了改性后的氰酸酯树脂在高低温(-196、200 ℃)环境下力学性能,分析其增韧机理和影响低温力学强度的关键因素,考察改性后氰酸酯树脂的耐热性、阻燃性和经高低温冷热循环(-196~200 ℃)后的耐久性等性能并给出结论。
1 实验部分
1.1 原料
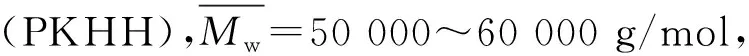
1.2 自制促进剂M
原材料使用前经过除水干燥处理,比例称取一定量的Oap-POSS和l,2-二氯乙烷,混合放置在装配好的反应容器内,干燥通氮气保护,升温至60~70 ℃,待体系成均匀透明液体时,缓慢滴加一定量的间甲基苯基异氰酸酯,维持反应5 h,反应完毕降到室温后处理,干燥得到略棕黄色固体(实物见图2),封存待用。
1.3 试样配比及制备
中温固化改性氰酸酯树脂各组分配比见表1。
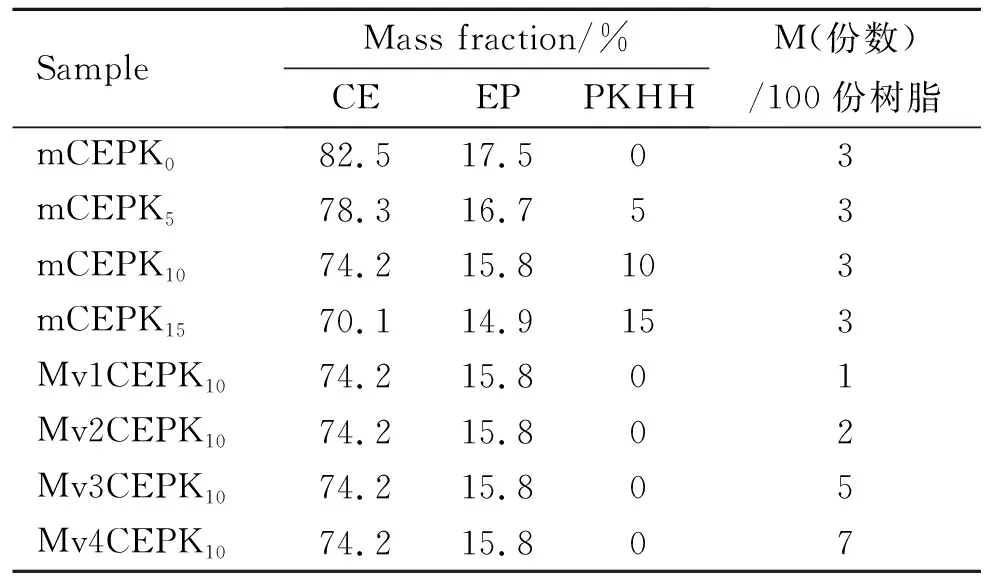
表1 中温固化改性氰酸酯树脂各组分配比
其中不同试样的PKHH含量不同,CE/EP的质量比维持不变。按照表1中配比要求(不含PKHH的除外),将PKHH加入到130 ℃的EP中,待完全溶解后将其加入已溶解的CE中,升温至(150±5)℃,高速搅拌(1000 ~1500 r/min),维持2~3 h,得到均一透明的液体,降温到90 ℃加入促进剂M,混合均匀即可将其倒入已预热好(120 ℃)的模具中,真空脱泡,按照125 ℃/4 h固化(未特殊说明,均按此工艺制度固化),得到四种不同配比的固化物分别为mCEPK0、mCEPK5、mCEPK10、mCEPK15和四种不同含量促进剂M的改性氰酸酯树脂,即Mv1CEPK10、Mv2CEPK10、Mv3CEPK10、Mv4CEPK10。
1.4 分析与测试
差示扫描量热分析(DSC):采用美国waters公司DSC2500型差示扫描量热分析仪测定反应热焓和固化曲线,升温速率10 ℃/min,扫描温度为40~400 ℃。固化度α计算方法见式(1):
(1)
式中α为固化度;ΔH为反应总热焓,J·g-1;ΔHr为反应剩余热焓,J·g-1。
红外光谱分析(FTIR):采用美国nicolet6700型傅立叶红外光谱仪,KBr压片法,扫描范围为4000~500 cm-1。
力学性能测试:采用美国INSTRON-5969型万能材料试验机和摆锤冲击试验机,按GB/T2567—2008相关测试方法对试样的拉伸性能、弯曲性能和冲击韧性进行测试。热老化测试是将拉伸试样放置在150 ℃老化箱中,经过500 h老化后,测定室温拉伸强度和模量。
动态热机械分析:采用日本精工DMS6100动态机械热分析仪,测试模式双悬臂,试样尺寸为50 mm×10 mm×3 mm,振动频率为1 Hz,扫描范围为-150~250 ℃,升温速率为5 ℃/min。
扫描电镜测试:对待测样品断面进行喷金处理,采用SU3500型扫描电子显微镜观察试样断面形貌。
热失重分析:采用日本精工6300型热失重分析仪测定试样的热分解特性,测试样品质量约5.0 mg,空气氛围,升温速率为10 ℃/min,温度范围50~700 ℃。
低温性能测试:测定树脂浇铸体试样在-196 ℃下的拉伸、弯曲强度和冲击韧性。抗拉强度、弯曲强度和冲击韧性的测试方法和试样尺寸均参照GB/T2567—2008的相关规定执行,将处理好的拉伸和弯曲试样放置在自制低温保温套件中,保温10 min后开始测试。冲击韧性测试:试样在装有液氮的自制保温套件中保温10 min后,取出5 s内完成测试。
冷热循环测试:试样尺寸为φ35 mm×5 mm。实验前将试样放置于50 ℃干燥箱中,恒温30 min,然后取出,迅速将其放入装有液氮的保温器皿中,保温10 min,取出放入200 ℃恒温箱内保温10 min,冷-热循环后测定试样表面的裂纹密度(根/cm2)。
核磁谱图测试:采用美国Varian的Unity 300 Spectrometer核磁共振仪对制备的促进剂M进行硅谱测试。测试频率300 MHz,CDCl3为溶剂,内标为TMS,加入少量的乙酰丙酮铬作为弛豫试剂。
耐空间环境测试:带电粒子辐照:辐照源(60 Co),总辐照剂量:3×103Gy;紫外辐照条件:环境压力(5.3×10-4Pa),辐照光谱(1×10-8~4×10-7m),太阳辐照光谱×总辐照量(1.17×109J/m2)。
阻燃测试:材料的阻燃性测试按照《美国阻燃材料标准》ANSI/UL-94-1985相关内容执行。
流变性能测试:采用英国Gemini200型流变仪对树脂的流变特性分析,平行板直径25 mm,板间距500 μm,升温范围:40~200 ℃,升温速率5 ℃/min,震荡模式,振动负荷频率为1 Hz。恒温流变测试温度选定为120 ℃和100 ℃。
2 结果与讨论
2.1 促进剂M的结构表征
图1为Oap-POSS改性前后的FTIR谱图。图2为合成促进剂M的29Si-NMR谱图和实物图。
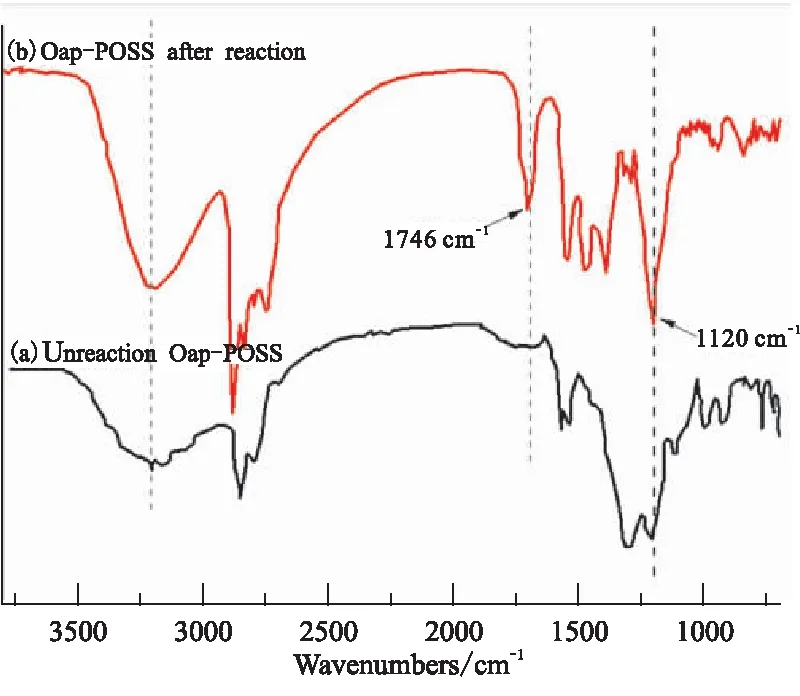
图1 Oap-POSS反应前、后的FTIR谱图
图2 合成的促进剂M的29Si核磁谱图和实物图
由图1看出,Oap-POSS与间甲基苯基异氰酸酯反应后,伯胺基特征峰移至3367 cm-1处,可认为是仲胺基特征峰。1746 cm-1处出现特征峰为碳氧羰基伸缩振动峰,1500 cm-1处苯环特征峰信号变强,并且1120 cm-1硅氧伸缩振动特征峰不变,分析结果证实反应产物中含有苯基脲结构,并且POSS中Si—O键结构未受影响。
由图2看出,反应产物仅含有一种硅氧结构POSS体,反应过程中Si—O键未被破坏,合成产物促进剂M为淡黄色固体,分子结构如图2中所示。因此,结合图1和图2结果,证实反应路线可行且产物为目标产物。
2.2 促进剂M对mCEPK树脂固化行为的影响
利用DSC法考察mCEPK树脂固化行为。首先选用固定配比的PKHH/EP改性氰酸酯树脂混合物(CEPK10)为基础物,通过加入不同含量促进剂M,考察其促进剂用量对mCEPK树脂固化行为的影响。图3为不同促进剂M含量的CEPK10树脂DSC曲线。
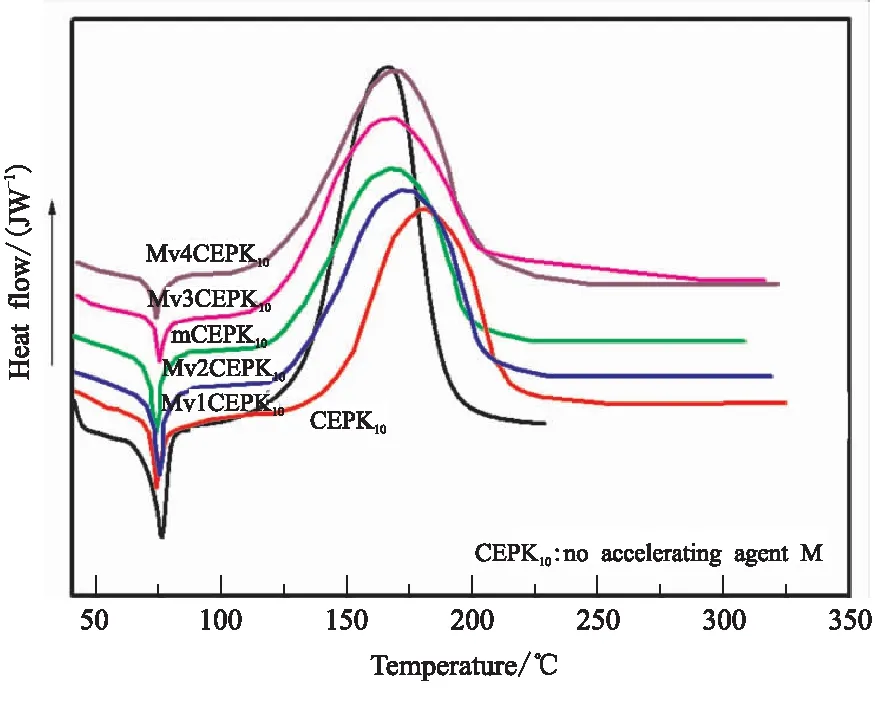
图3 不同促进剂M含量的CEPK10树脂DSC曲线
由图3可知,加入促进剂M的CEPK树脂混合物(Mv1CEPK10)的起始反应温度相比不含促进剂M的树脂(CEPK10)降低约50 ℃,且随着促进剂M含量的增加,CEPK10反应放热峰进一步向低温区移动,含有3%促进剂M的CEPK10树脂体系(mCEPK10)的起始反应温度降至约119.9 ℃,随着促进剂M的进一步增加,CEPK10树脂体系的起始反应温度变化不明显,可见过多的促进剂M对降低CEPK树脂体系固化温度没有明显效果,考虑到促进剂M的加入可能影响树脂体系的贮存期,本文选择mCEPK树脂体系的促进剂M添加量为3%。进一步看出加入促进剂M后CEPK10反应放热峰变宽,说明在促进剂M的存在下CEPK树脂体系固化反应平缓且达到较完全固化程度需要延长反应,因此本文选用125 ℃/4 h为mCEPK树脂体系的固化工艺。
其次固定促进剂M的含量(3%),改变CEPK树脂中PKHH的含量,考察PKHH的含量对mCEPK固化行为的影响。图4为相同促进剂M含量的mCEPK树脂DSC曲线。由图4看出,随着PKHH的增加mCEPK树脂体系的反应峰值温度向低温移动,DSC曲线形状相似,插图中固化后mCEPK树脂DSC曲线无峰形出现(250 ℃之前),这说明PKHH对mCEPK树脂体系固化有协同促进作用,改性后mCEPK树脂体系经125 ℃/4 h后固化较完全且不改变反应路径。
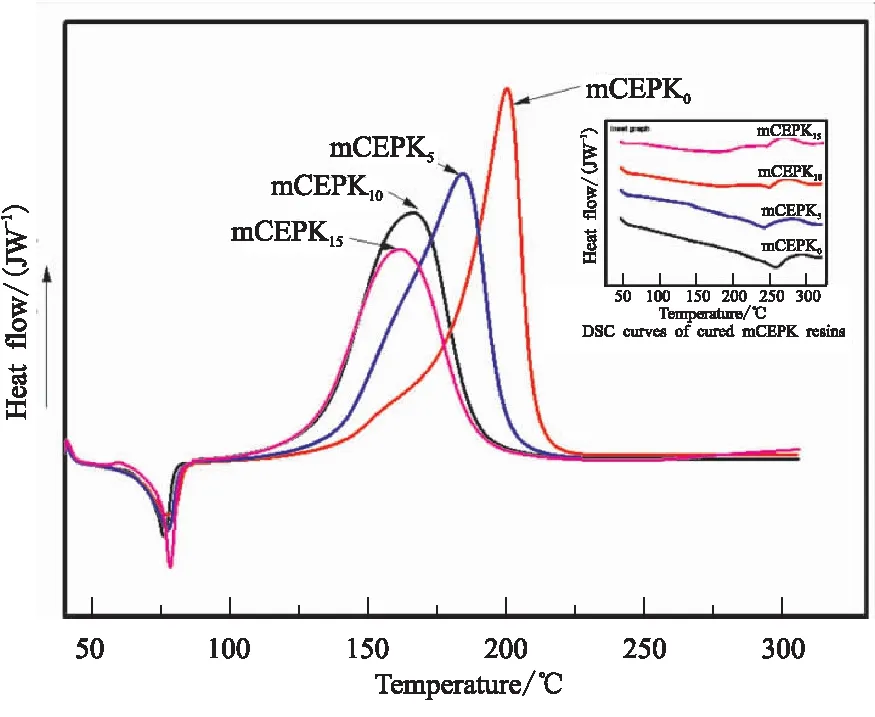
图4 不同PKHH含量的mCEPK树脂DSC曲线
表2为不同PKHH含量的mCEPK树脂混合物反应参数表。表2数据结果进一步证明,经规定的固化工艺固化后,含有PKHH的试样的固化度α明显提高,且随着PKHH含量的增加固化度进一步提高,其中mCEPK15固化度接近97%。因此,综合上述分析结果可推断出促进剂M和PKHH对氰酸酯树脂的固化有较好促进作用,选定的固化工艺合理。

表2 不同PKHH含量改性mCEPK树脂混合物固化反应参数表
2.3 mCEPK树脂流变行为
通过测定mCEPK树脂体系的黏度随温度、时间变化规律,考察了树脂体系的流变行为和工艺适应性。图5为mCEPK树脂的流变曲线,其中图5(a)mCEPK树脂的变温流变曲线;(b)mCEPK10树脂的等温流变曲线。由图5(a)看出,mCEPK树脂的最低黏度平台出现在约120~130 ℃范围内,随着树脂中PKHH含量的增加树脂体系的最黏粘度成指数倍增加(409~6500 cp),最低粘度平台范围缩短且最低粘度激增时的温度从138.8 ℃降到约130 ℃,而不含有促进剂M的CEPK5树脂最低黏度激增时温度为171 ℃且平台期较长,这种现象是由促进剂M和PKHH共同作用的结果同时证实了DSC的分析结论。分析图5(b),mCEPK10125 ℃等温条件下,体系黏度随时间成指数级增长,30 min内树脂体系黏度增加约1000倍,到达初步固化状态,可说明本文选定的固化工艺合理。100 ℃等温条件下,树脂黏度增长速率减慢,40 min中内黏度基本不变,维持在2000 cp左右,上述结果可以推断出,树脂体系粘度平台期时间随温度的降低而延长。因此,通过对mCEPK树脂流变行为的分析可知,对于实际施工制备复合材料构件而言mCEPK树脂体系具有适中的工艺操作窗口。
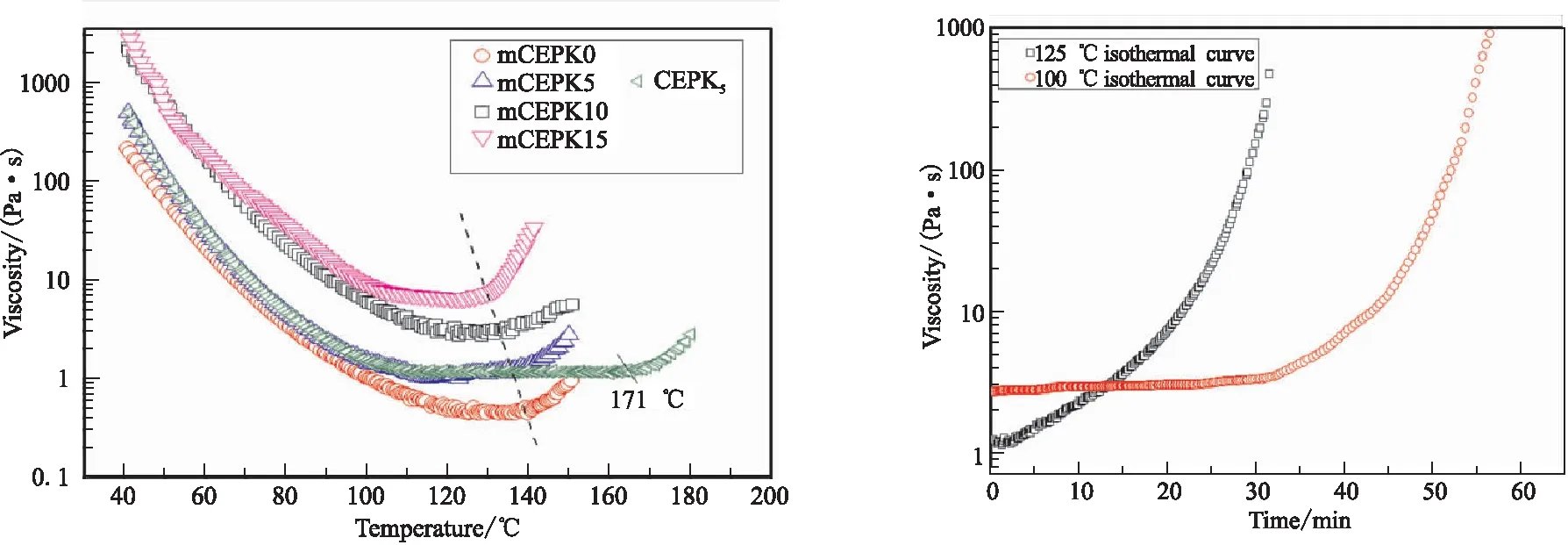
( Rheological curves in heterotherm (b) Isothermal rheological curves
2.4 mCEPK树脂的增韧机理
mCEPK树脂固化物的冲击断面微观形貌如图6所示,其中(a)、(b)为mCEPK0和mCEPK1524 ℃下冲击断面微观形貌图,(c)、(d)为-196 ℃下冲击断面微观形貌图。冲击数值结果见表3。

表3 PKHH改性mCEPK树脂的冲击韧性
由图6(a)、(b)看出,24 ℃下随着PKHH的增加,试样断面粗糙度增加,裂纹扩展被束缚并出现弯曲和分叉,呈现韧性断裂。表明PKHH的加入能明显提高CE/EP混合物抵抗外载荷的能力。与传统的塑料、橡胶增韧氰酸酯树脂不同,mCEPK树脂中并没有出现相分离球状形态,这说明线性的PKHH长链分子在CE/EP固化网络中有较好的相容性。因此,推断PKHH与CE/EP混合物可能形成半互穿网络结构(SIPN)[11-13],可能的解释为由于PKHH的多羟基结构与CE树脂极性相近,通过PKHH长链分子链缠绕的“协同效应”和“强迫包容”作用提高了CE/EP混合物的韧性[14]。

(24 ℃, mCEPK0 (b) 24 ℃, mCEPK15
由图6(c)、(d)看出,在-196 ℃条件下,mCEPK0试样断面出现河床状断裂纹路,这可能是超低温下分子间的堆积更加紧密,分子间作用力加大,因此表现出了一定的韧性断裂特征。同时,随着PKHH的增加裂纹尖端成分叉形态且韧性进一步提高,这是由于PKHH的多醚键结构超低温下具有较好的活动能力且抵消外载荷作用,因而体现出mCEPK15树脂更好的低温韧性。
通过DMA法进一步研究了mCEPK树脂的固化网络构成和韧性关系。图7为不同PKHH含量的mCEPK树脂固化物的损耗模量曲线。由图7可知,mCEPK树脂中随着PKHH的增多,网络主结构α峰和链段次级β峰均有向低温移动的趋势且β峰的峰宽变大。可能的解释如下:(1)PKHH存在导致CE树脂交联点间的距离增加和内聚强度降低,因此体现出α峰向低温转移;(2)PKHH的多醚键结构使得mCEPK树脂固化网络中可具备超低温下活动的基团、链节数增多,导致了β峰向更低温度区域移动;(3)尽管PKHH的玻璃化温度与CE树脂的相差较大,由于PKHH自身多羟基的特点,CE树脂固化网络对其产生了“强迫包容”作用,仅展现出了有一个α峰。综合上述结果,发现PKHH的加入从微观结构上提升了CE/EP固化物的在高温和超低温环境下抗外载荷的能力,体现出高低温冲击韧性均高于纯CE树脂。
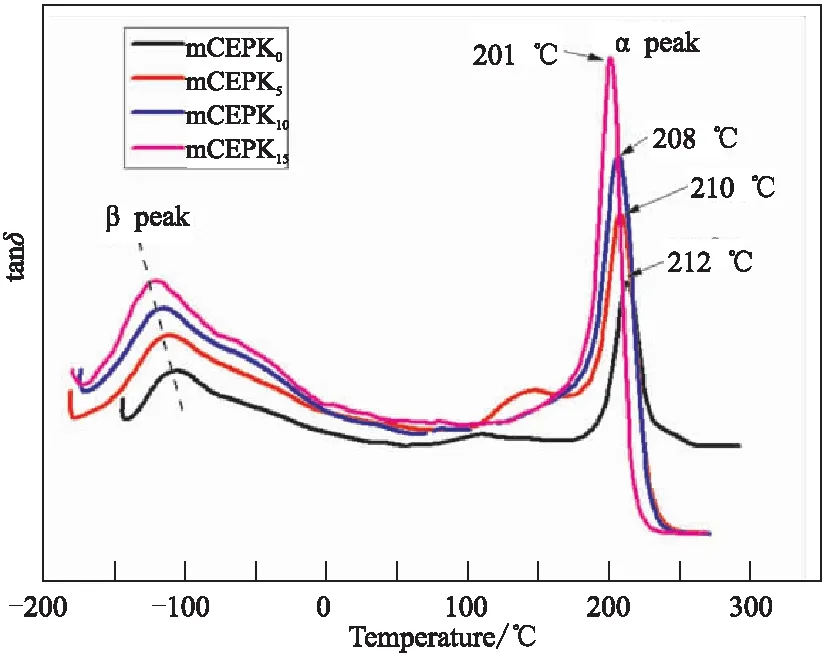
图7 不同PKHH含量mCEPK树脂固化物的DMA曲线
2.5 mCEPK树脂的综合性能
采用本文规定的固化工艺对mCEPK树脂进行固化,测定固化后树脂的力学性能、热稳定性和阻燃性等性能,所得数据列于表4。结合表4数据总结了PKHH的加入mCEPK树脂固化物力学性能、热稳定性和阻燃性等性能影响因素。

表4 mCEPK树脂固化物力学性能、热稳定性和阻燃性数据表
由表4看出,中温固化后mCEPK改性氰酸酯树脂拉伸强度超过60 MPa,模量超过3.0 GPa,具有较好的力学强度和模量,满足航天复材构件应用条件。超低温条件下,树脂的强度和模量均提高约20%,这结果与2.2节分析的一致。随着PKHH加入对CE树脂的热失重温度略有降低,但是对整个固化网络的热稳定性影响不大,PKHH加入后的mCEPK树脂的阻燃性好于传统改性环氧阻燃性且与纯CE树脂相当。进一步由表4数据看出,PKHH的加入对CE树脂固化物的线膨胀系数α影响不大,平均值介于(30~40)×10-6℃-1之间,比一般改性环氧树脂的线膨胀系数降低约50%,有利于树脂固化物在冷热循环中长时间应用。由此可见,适量的PKHH含量的增加有效改善CE固化物的缺陷,提高力学性能,热稳定性、阻燃性和线膨胀系数与纯CE树脂相当。
2.6 mCEPK树脂的耐久性能
测定mCEPK0、mCEPK5、mCEPK10、mCEPK15试样的高温力学强度和高温老化性能测试,考察mCEPK树脂在实际应用环境中的高温耐久性。表5为mCEPK树脂固化物不同温度下的拉伸性能,图8为mCEPK树脂固化物经热老化后的拉伸性能。
由表5可知,随着PKHH增加mCEPK固化物在150 ℃的强度基本相同,强度保持率均大于85%,同时发现200 ℃强度下降较多,强度保持率仅有30%~40%,且数值变化无规律,这主要是因为200 ℃测试条件接近混合物的玻璃化温度,导致网络分子活动能力增加,抵抗外载荷的能力下降。由图8可知,经过150 ℃热老化100 h后,mCEPK固化物强度保持率达到90%以上。综合分析,在玻璃化转变温度以下50 ℃附近,PKHH改性CE/EP固化物具有较好的热稳定性和耐久性,含有5%~10%的PKHH的固化物在200 ℃具有较高的强度。
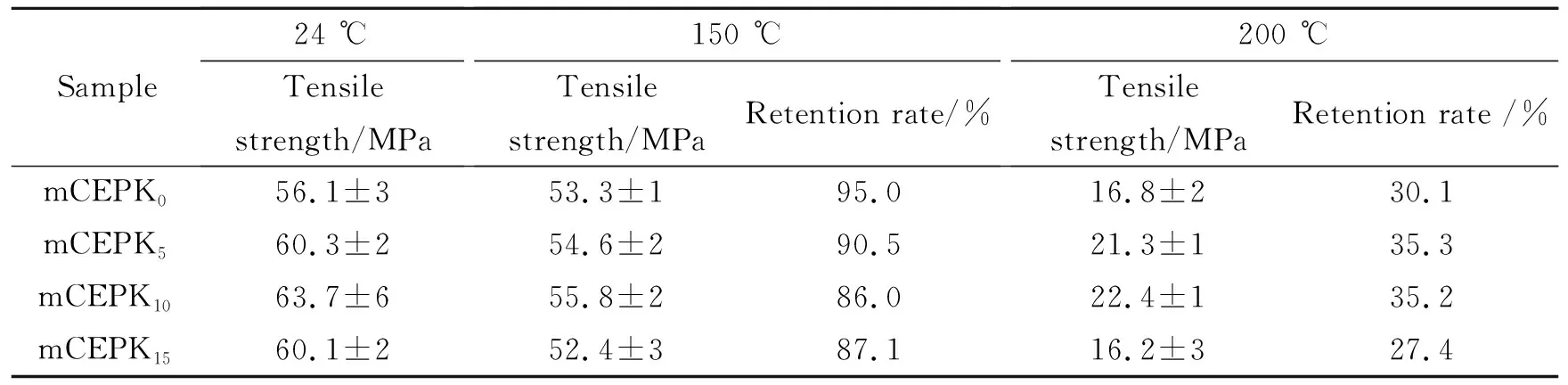
表5 mCEPK树脂不同温度下的拉伸性能
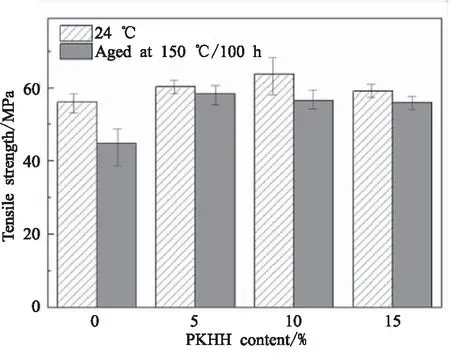
图8 不同PKHH含量mCEPK固化试样24 ℃和150 ℃热老化500 h后抗拉性能
测定mCEPK树脂经冷热循环(-196~200 ℃)后,试样表面外观形貌,考察树脂体系在航天超低温环境中耐久应用的适用性。表6为不同树脂固化物经冷热循环后材料首次出现裂纹情况。图9为mCEPK10试样经冷热循环30次后的形貌。为了对比本实验同时制备CE/EP/聚砜/促进剂M(用相同份数的聚砜(PSF)取代PKHH)试样和纯CE树脂/促进剂M试样。由表6看出,适当含量PKHH的加入有效改善了CE树脂的耐冷-热交变性能,过多的PKHH加入对CE树脂的耐低温稳定性提升不明显,但是纯CE和聚砜改性CE/EP后树脂固化物的冷热循环稳定性变差明显,主要是由于PSF在CE树脂中分散相态和CE自身脆性引起的。另外,由图9看出,mCEPK10试样低温破坏的裂纹出现弯曲、分叉等现象,说明PKHH的加入有效的改善了CE树脂的低温稳定性。
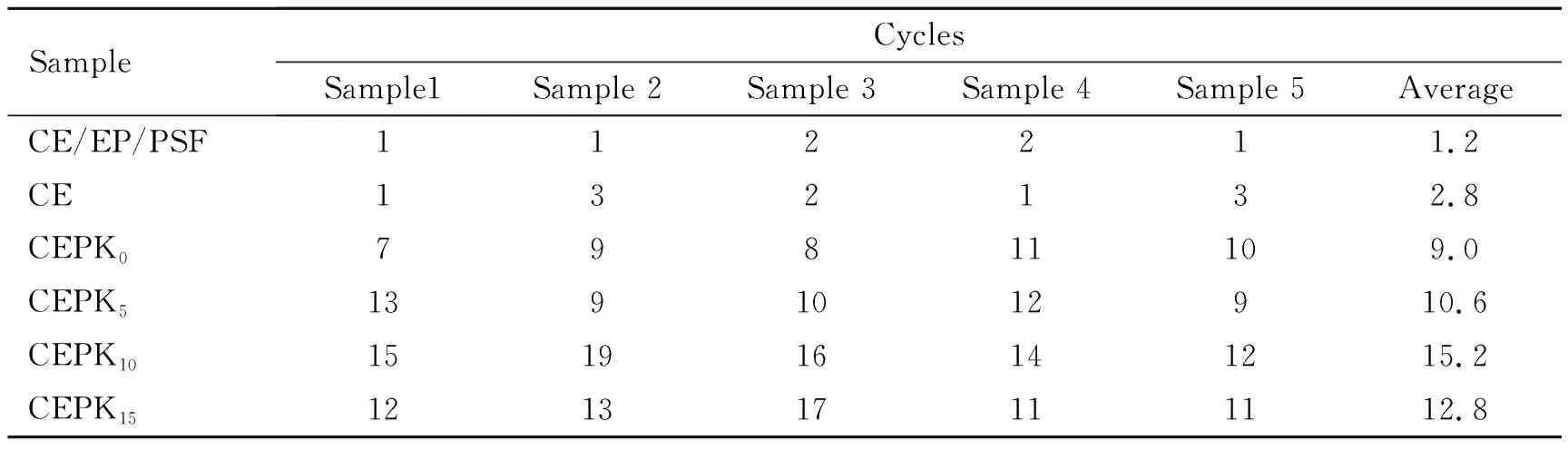
表6 不同树脂固化物经多次冷、热循环后表面首次出现裂纹情况

( Sample before the test (b)Sample after the test
2.7 mCEPK树脂的空间性能
选择综合性能较好的mCEPK10树脂测试其拉伸试样经电子辐照和紫外辐照后的强度变化数据,考察mCEPK树脂的耐空间环境性能。表7为mCEPK10树脂经电子辐照和紫外辐照后拉伸强度数据表。由表7可见,空间电子辐照和紫外辐照会对树脂结构强度和网络分子构成有一定影响并且影响的程度随温度升高而加大,mCEPK10经过辐照后室温、150 ℃测试强度保持率均超过80%,说明mCEPK树脂的空间耐辐照适应能力较好。
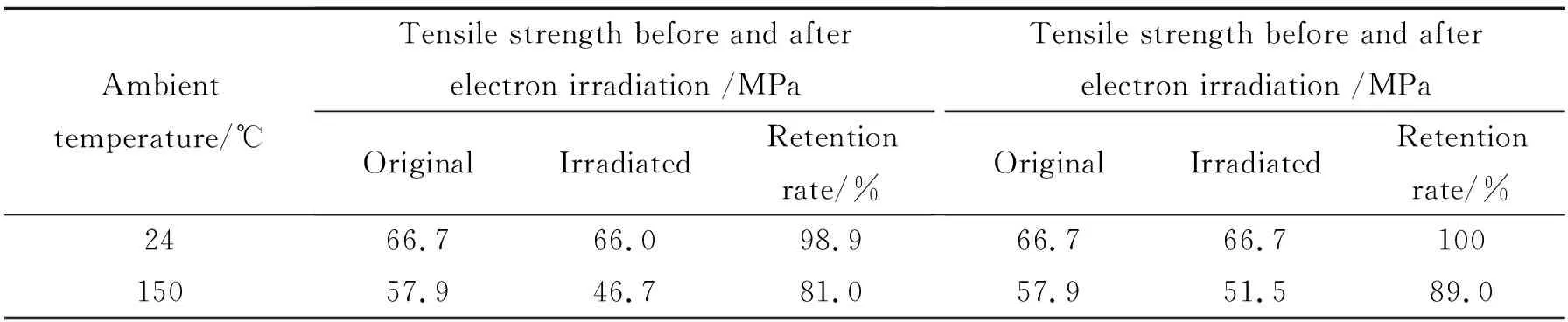
表7 mCEPK10树脂经电子辐照和紫外辐照后拉伸强度数据
3 结论
(1)采用POSS体与间甲基苯基异氰酸酯反应制备了含有纳米基团的取代脲衍生物,合成产物为目标产物促进剂M。促进剂M可实现改性CE树脂125 ℃下固化且固化度超过90%,室温贮存期超过15 d,改性CE树脂100 ℃/40 min内维持2000 cp黏度不变。
(2)PKHH和EP树脂共改性CE树脂并混合分散加入促进剂M,获得了兼具良好低温力学强度、优异的冷-热循环耐久性、适宜的工艺性和良好的空间环境适应性的中温固化改性氰酸酯树脂mCEPK。
(3)PKHH能有效地提高CE树脂的耐低温性能,10%PKHH的加入可使CE树脂的-196 ℃冲击强度提高约40%,并且对CE/EP树脂的模量影响较小,10%PKHH的加入可使CE树脂经过冷、热循环首次出现裂纹的次数提高近5倍。
猜你喜欢
建材发展导向(2021年19期)2021-12-06
建材发展导向(2021年7期)2021-07-16
宇航材料工艺(2021年2期)2021-05-15
阅读(快乐英语高年级)(2021年11期)2021-03-08
建材发展导向(2020年16期)2020-09-25
科学导报·学术(2019年19期)2019-09-10
读者·校园版(2018年5期)2018-02-08
山东工业技术(2017年20期)2017-10-17
科学与财富(2017年22期)2017-09-10
科学与财富(2017年18期)2017-07-09
