
代谢工程改造大肠杆菌MG1655积累L-苹果酸
2020-10-29 02:30:06周志东
武汉科技大学学报 2020年6期
何 彬,周志东,曹 阳,黄 皓,周 卫,陈 俊
(武汉科技大学化学与化工学院,湖北 武汉,430081)
L-苹果酸是生物体三羧酸(TCA)循环中的一种重要中间产物,广泛应用于食品、医药、化工等多个领域[1]。早期生产苹果酸的方法主要有化工合成法、植物提取法以及采用琥珀酸为原料的酶转化法[2],然而随着此类方法所需成本日益增加、人们的环保意识不断增强,节能、环保且高效的微生物发酵技术已成为获取苹果酸的重要途径。作为已知的苹果酸高效生产菌株,曲霉属菌种诸如黄曲霉、黑曲霉和米曲霉等均能以葡萄糖为碳源积累苹果酸,尤其使用黄曲霉获取L-苹果酸最大积累量可达100 g/L,但是在其发酵过程中通常伴有曲霉毒素产生,不适合用于大规模工业生产[3-6],因此,研究者开始尝试使用其它菌株发酵生产L-苹果酸,如刘亚等[7]利用米根霉突变菌株,通过初步优化发酵工艺,以葡萄糖为碳源,经72 h发酵,所得L-苹果酸积累量达到了57.71 g/L。同时,随着基因工程的发展,通过基因编辑得到基因工程菌发酵生产L-苹果酸的技术日渐成熟,娄菲等[8]通过敲除大肠杆菌E21中的苹果酸酶基因maeA和maeB,使得苹果酸产量提高了36%;陈修来等[9]通过在酿酒酵母中表达来自黄曲霉的丙酮酸羧化酶、苹果酸脱氢酶以及C4-二羧酸转运蛋白,成功构建了L-苹果酸的合成路径,使得工程菌能够积累L-苹果酸,为相关合成技术提供了参考;Dong等[10]通过在大肠杆菌w3110中过表达pos5基因,增加了NADPH含量,使得最终的L-苹果酸产量达到9.34 g/L。虽然可通过在大肠杆菌或酿酒酵母等模式菌中过表达异源的苹果酸酶基因来提高L-苹果酸产量[11],但因为不涉及菌株基因组的改造,其性状通常难以稳定遗传。另一方面,基因编辑技术的快速进步也为代谢工程改造菌株提供了动力,如Ⅱ型CRISPR系统最初被发现是细菌和古细菌用来防止外源DNA入侵的一种免疫系统[12-15],相对于传统的同源重组技术,Ⅱ型的CRISPR/Cas9基因编辑系统具有效率高、周期短、操作步骤简单等特点,对单个基因的编辑效率最高可达100%[16]。基于此,本文选取遗传背景清楚且易于进行分子操作的大肠杆菌MG1655,借助CRISPR/Cas9基因编辑系统对其进行代谢工程改造来积累L-苹果酸,以期为微生物发酵技术生产L-苹果酸提供参考。
1 试验
1.1 材料及基因改造
1.1.1 培养基
LB液体培养基中胰蛋白胨、酵母提取物及氯化钠的质量浓度分别为10、5、10 g/L,另外,在此基础上按15 g/L添加琼脂粉制得LB固体培养基。
M9培养基中Na2HPO4、KH2PO4、NaCl、NH4Cl及葡萄糖的质量浓度分别为6.78、3、0.5、1、20 g/L,且在使用时按1%的体积分数添加适量的MgSO4和CaCl2。
1.1.2 酶和试剂
质粒DNA提取试剂盒、琼脂糖凝胶DNA提取试剂盒为美国OMEGA公司产品;DNA Marker、2×ES Taq MasterMix为南京诺唯赞生物科技股份有限公司产品;苹果酸、丙酮酸、乳酸、乙酸等有机酸标准样品为Sigma-Aldrich公司产品;其它试剂均为国产分析纯试剂。
1.1.3 CRISPR/Cas9介导的基因敲除
本研究所用野生型大肠杆菌MG1655和大肠杆菌DH5α均为本实验室所保存,质粒pTargetF和pCas购于中国科学院上海生命科学研究院,重组质粒和基因敲除菌株均由本实验室构建。基因改造后的代谢途径如图1所示,在利用CRISPR/Cas9基因编辑系统对大肠杆菌MG1655进行基因敲除之前,需进行敲除载体pTargetF-sgRNA的构建及同源臂片段的制备。

图1 大肠杆菌MG1655部分代谢流程图
在构建poxB基因敲除载体时,以pTargetF质粒做模板,以poxB-P1和poxB-P2为引物,通过全质粒PCR扩增,得到含有poxB 相关sgRNA的pTargetF,扩增条件为:预变性95 ℃,300 s; 95 ℃,30 s; 58 ℃,30 s;72 ℃,90 s,32个循环; 72 ℃,600 s,4 ℃保存。使用DpnⅠ酶进行消化,消除pTargetF原始模板,转化大肠杆菌DH5α,提取重组质粒,用引物maeA-P3和maeA-P4借助美国UVP公司GelDoc-It310型凝胶成像仪进行PCR鉴定[13],所构建的重组质粒命名为pTargetF-poxB。按同样的方法,分别构建pta-ackA、ldhA、pflB、maeA、maeB和pfkA基因的敲除载体。
在制备poxB同源臂片段时,以大肠杆菌MG1655基因组为模板,以引物poxB-P5和poxB-P6扩增poxB基因的上游同源臂,以引物poxB-P7和poxB-P8扩增poxB基因的下游同源臂。然后以poxB-P5和poxB-P8为引物,以上、下游同源臂做模板,通过SOE-PCR进行扩增,得到上、下游同源臂的融合片段[15]。按同样方法,利用不同引物分别制备pta-ackA、ldhA、pflB、maeA、maeB和pfkA基因上、下游同源臂的融合片段。
在敲除载体构建完毕并制得同源臂片段后,利用CRISPR/Cas9基因编辑系统对大肠杆菌MG1655进行基因敲除。首先敲除其poxB基因,获得M01菌株,具体敲除步骤如下:1)将pCas质粒转化至大肠杆菌MG1655中,获得含有pCas 质粒的大肠杆菌MG1655菌株(卡那霉素抗性);2)挑取单菌落接种到含有50 mg/L卡那霉素的LB液体培养基中过夜培养,之后以1%的接种量转接到25 mL卡那霉素质量浓度为50 mg/L的 LB液体培养基中,加入终浓度为10 mmol/L的阿拉伯糖诱导pCas质粒上red蛋白的表达,在30 ℃的温度下,以200 r/min转速振荡培养至OD600约为0.4左右时,再利用美国贝克曼库尔特公司J-26XP型高速冷冻离心机冷冻离心回收菌体并制作用于电转化的感受态细胞;3)利用BIO-RAD公司165-2100型电转化仪进行电转化处理。向50 μL感受态细胞中加入100 ng pTargetF-poxB重组质粒和100 ng目的基因同源片段,混匀后加入经预冷处理、直径为1 mm的电转杯中,在1.8 kV电压下进行电转,然后迅速转入预冷的1 mL的LB液体培养基中,在30 ℃的温度下,以200 r/min转速培养1 h以复苏细胞;4)最后将菌体接种于含有50 mg/L卡那霉素和50 mg/L壮观霉素的双抗性LB固体培养基上,在30 ℃的温度下,以200 r/min转速培养过夜,获得M01菌株,回收菌体,使用细菌DNA提取试剂盒提取基因组,利用引物P5/P8进行PCR鉴定[17-18]。按照同样方法,在M01菌株基础上继续敲除pta-ackA、ldhA、pflB等基因,依次获得M02、M03和M04菌株,阻断相关副产物的合成以积累丙酮酸,增大TCA循环的通量;然后在M04菌株基础上继续敲除maeA和maeB基因以防止苹果酸的消耗[19],所得菌株依次为M05和M06;最后再敲除M06菌株的pfkA基因得到M07菌株,确保葡萄糖尽可能通过磷酸戊糖途径代谢,从而产生更多的辅酶NADPH,为合成苹果酸提供还原力[20]。本研究中所用重组质粒及所制基因敲除菌株列于表1,所用引物列于表2。需要指出的是,每一次基因敲除成功后所得菌株会同时含有pCas质粒和pTargetF重组质粒,为了消除这两个质粒,首先将菌株接种至含有50 mg/L卡那霉素和终浓度为1.0 mmol/L IPTG的LB液体培养基中,在30 ℃的温度下,以200 r/min转速培养8~16 h,再将其涂布于含有50 mg/L卡那霉素的LB固体培养基上,挑取单菌落,检测其壮观霉素抗性,如无壮观霉素抗性,则表明pTargetF重组质粒已消除,继续挑取该单菌落接种于不含抗性的LB液体培养基中,在37 ℃的温度下,以200 r/min转速培养8~16 h后涂布无抗性LB固体培养基,挑取单菌落检测其卡那霉素抗性,如无卡那霉素抗性,则表明pCas已消除。

表1 菌株和质粒Table 1 Strains and plasmids

表2 引物Table 2 Primers

续表2
1.2 发酵处理
对野生型大肠杆菌MG1655及M04、M05、M06、M07菌株分别进行有氧摇瓶发酵处理。将-80 ℃保存的菌株在添加相关抗生素的LB平板上划线,挑取单菌落接种到装有5 mL液体LB培养基的试管中,在37 ℃温度下以200 r/min的转速培养过夜,再以1%的接种量转接到装有50 mL液体M9培养基的锥形瓶中,在37 ℃温度下以200 r/min的转速发酵处理48 h。
在好氧发酵条件下,以葡萄糖为底物,通过糖酵解或磷酸戊糖途径生成丙酮酸,之后经过TCA循环氧化生成L-苹果酸,通过这一途径,1摩尔葡萄糖可以合成1摩尔L-苹果酸并释放出2摩尔的CO2,最大理论得率为100%,得率η的计算公式为
η=[n(生成的L-苹果酸)/n(初始葡萄糖)]
×100%
(1)
1.3 细胞代谢及产物分析
使用UV-2000型紫外分光光度计检测细胞生长量,检测波长为600 nm;利用P680型高效液相色谱仪检测发酵液中有机酸,分析条件为:色谱柱为AcclaimTM120C18(4.6 mm×250 mm,5 μm),流动相为0.1 mol/L的KH2PO4,用H3PO4调节液体pH为2.8,进样量为20 μL,紫外检测波长为215 nm,柱温为20 ℃,流速为1 mL/min。
2 结果与分析
2.1 敲除载体pTargetF-sgRNA的构建与鉴定
各个基因敲除载体pTarget-sgRNA的构建及验证结果如图2所示,其中图2(a)中泳道1到泳道6及图2(b)中泳道1分别为poxB、pta-ackA、ldhA、pflB、maeA、maeB、pfkA敲除载体的pTargetF-sgRNA全质粒PCR检测结果,上述质粒片段大小均与pTargetF相应值接近,约2000 bp左右;图2(c)中泳道1到泳道6及图2(d)中泳道1分别为poxB、pta-ackA、ldhA、pflB、maeA、maeB、pfkA敲除载体的pTargetF-sgRNA全质粒PCR鉴定结果,检测上游引物来自于pTargetF原始载体,下游引物来自各个基因相对应的20 bp sgRNA,上下游引物之间片段大小为536 bp。由检测结果可知,扩增片段大小与预期相符。对经PCR鉴定的敲除载体进行测序的结果表明,含20 bp目的基因的片段已成功克隆至pTargetF载体。
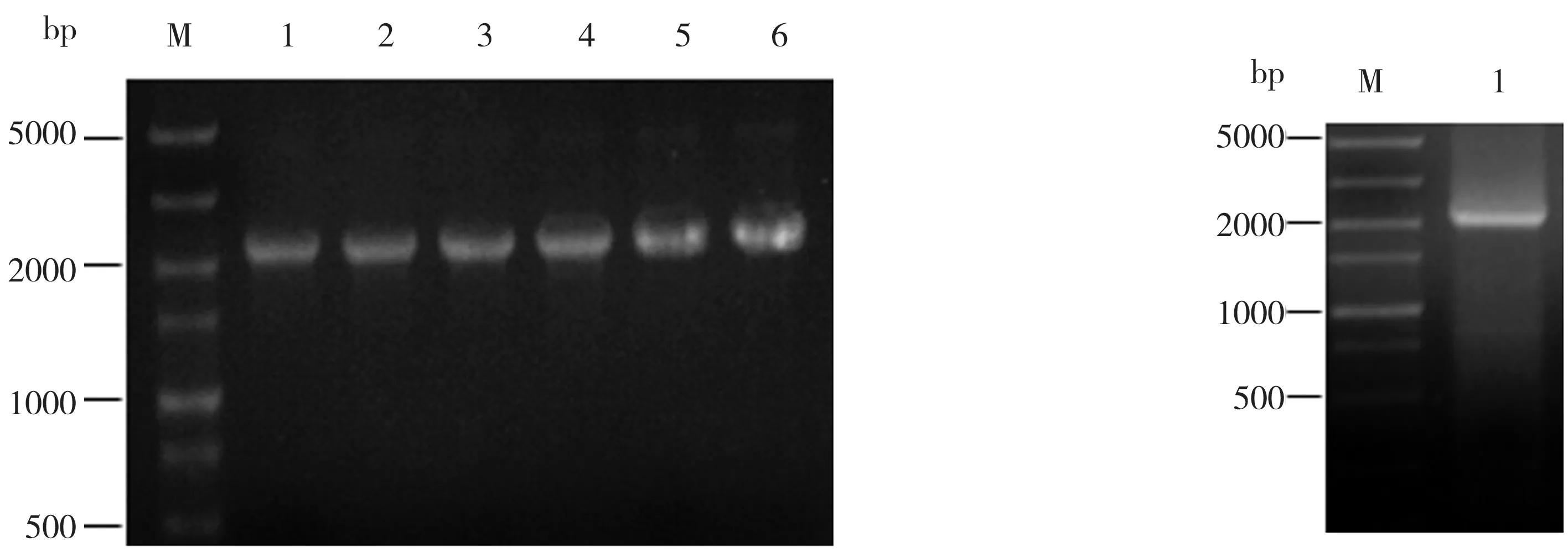
(a)poxB、pta-ackA、ldhA等的检测结果 (b)pfkA的检测结果
(c)poxB、pta-ackA、ldhA等的鉴定结果 (d)pfkA的鉴定结果
2.2 上下游同源臂的制备结果与分析
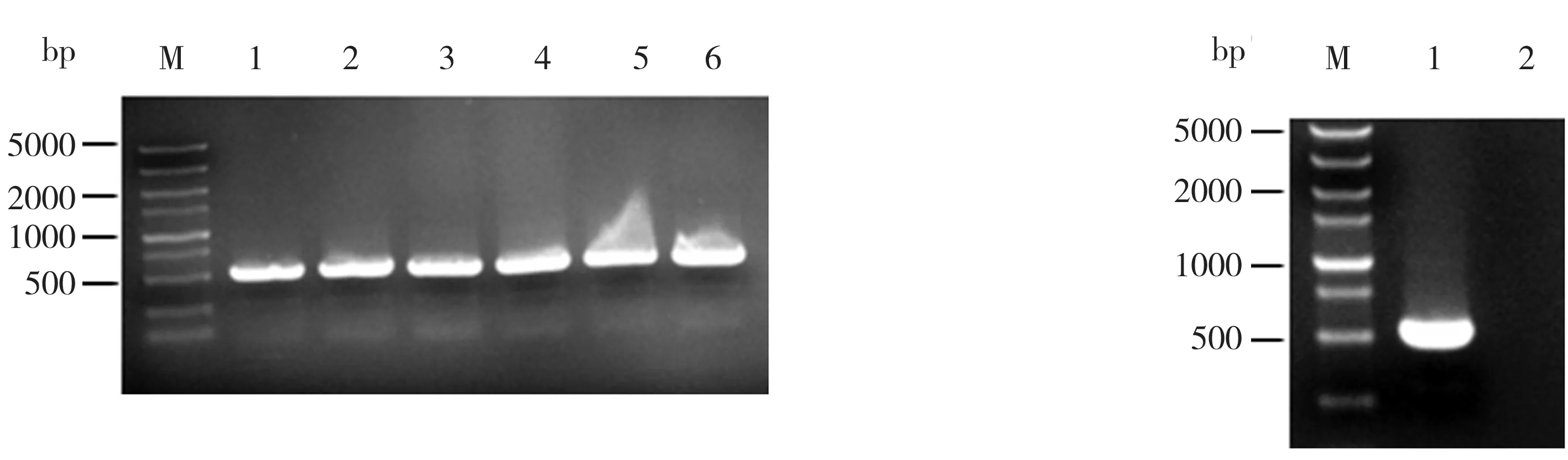
敲除基因的上下游融合同源臂片段的制备结果如图3所示,其中图3(a)中泳道1到泳道6及图3(b)中泳道1分别是poxB、pta-ackA、ldhA、pflB、maeA、maeB、pfkA基因的上、下游同源臂的融合片段。由图3可知,上述同源臂片段大小约为1000 bp,与预期一致,表明同源片段融合成功。

(a)poxB、pta-ackA、ldhA等的同源臂 (b)pfkA的同源臂
2.3 基因敲除菌株的构建结果与分析
基因敲除菌株利用引物P5/P8进行PCR鉴定的结果见图4,其中图4(a)中1泳道到6泳道为以野生型大肠杆菌MG1655基因组DNA为模板并分别以poxB、pflB、maeB、ldhA、maeA、pta-ackA的P5/P8为引物的PCR鉴定结果,7泳道到12泳道为以重组菌株大肠杆菌基因组DNA为模板并分别以poxB、pflB、maeB、ldhA、maeA、pta-ackA的P5/P8为引物的PCR鉴定结果;图4(b)中第1泳道为以野生型大肠杆菌MG1655基因组DNA为模板并以pfkA的P5/P8为引物的PCR鉴定结果,第2泳道为以重组菌株大肠杆菌基因组DNA为模板并以pfkA的P5/P8为引物的PCR鉴定结果。因为poxB、pflB、maeB、ldhA、maeA、pta-ackA、pfkA等基因的片段大小分别为1719、2284、2280、990、1698、3421、963 bp,基因上下游同源臂的融合片段大小约为1000 bp,若上述基因没有敲除成功,相应扩增的片段大小应分别为2719、3284、3280、1990、2698、4421、1963 bp,即上下游同源臂加上基因本身片段大小;若基因敲除成功,扩增的片段大小均为1000 bp左右,即仅仅是上下游同源臂融合片段的大小。由图4可知,图4(a)中1泳道到6泳道及图4(b)中1泳道所示poxB、pflB、maeB、ldhA、maeA、pta-ackA、pfkA基因敲除前扩增片段大小与图4(a)中7泳道到12泳道及图4(b)中2泳道所示上述基因敲除后的相应片段大小之差正好与所敲除基因片段大小相同,这与预期相符,可以判断相关基因已被成功敲除。
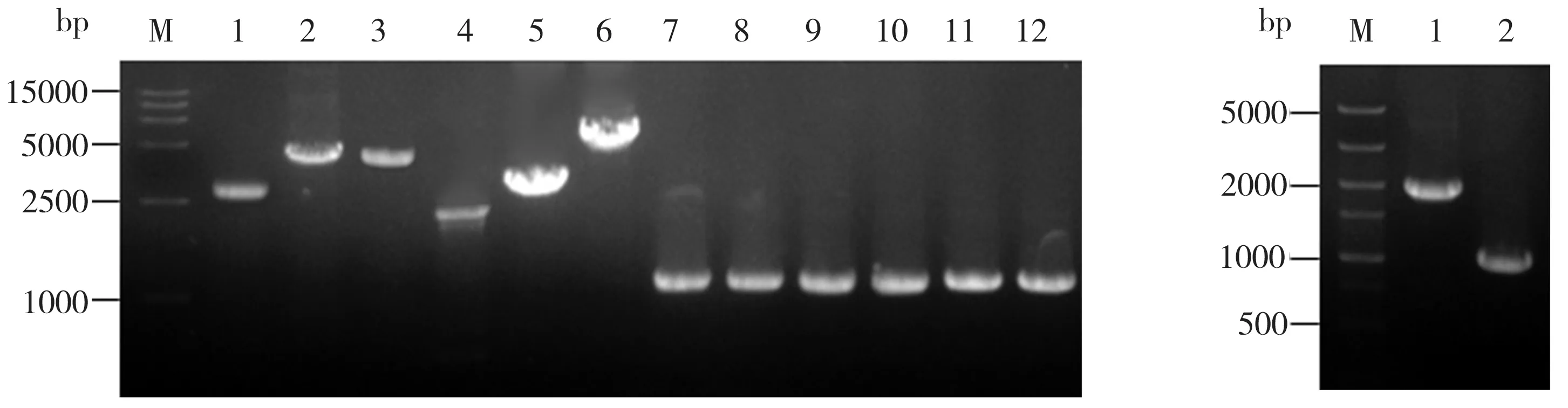
(a)以poxB、pflB、maeB等的P5/P8为引物 (b)以pfkA的P5/P8为引物
2.4 发酵液中有机酸分析
将野生型大肠杆菌MG1655及M04、M05、M06、M07菌株分别进行发酵处理,在摇瓶发酵48 h时对发酵液中的丙酮酸、L-苹果酸、乳酸和乙酸进行了测定,结果如图5所示。

图5 发酵液中有机酸的检测
从图5中可以看出,对比野生型大肠杆菌MG1655菌株和菌株M04发酵处理后的发酵液检测结果可以发现,野生型大肠杆菌MG1655菌株发酵后生成了较多的乳酸和乙酸等副产物,丙酮酸和L-苹果酸的积累量分别只有0.675、0.812g/L,而菌株M04发酵液中乳酸和乙酸积累量大量减少,丙酮酸和L-苹果酸的积累量分别达到5.621、4.012 g/L。这是因为菌株M04被敲除了丙酮酸副产物代谢途径上的4个基因,造成生成副产物的代谢途径被阻断,从而丙酮酸得到了大量积累,同时,丙酮酸的积累又促进了TCA循环的加速,从而导致TCA循环中对应的中间产物如L-苹果酸的积累量也相应增加。在菌株M04基础上敲除基因maeA后,所得菌株M05经发酵处理后,发酵液中L-苹果酸的积累量进一步增加至7.783 g/L,而丙酮酸的积累量则降低至4.016 g/L,这表明缺少苹果酸酶maeA催化后,L-苹果酸生成丙酮酸这一途径得到了有效的抑制,L-苹果酸的流失明显减少。而对于进一步敲除了苹果酸酶maeB的菌株M06来说,其发酵液检测结果显示L-苹果酸的积累量相比M05相应值并没有明显的增加,这应该是此时菌株胞内NAD(P)H/NAD(P)+值过低,还原力不足所致。相比菌株M06发酵检测结果,菌株M07发酵液中的丙酮酸积累量有所下降,但L-苹果酸积累量明显增加,达到了9.893 g/L,实际得率为66%,这是因为敲除菌株M06的pfkA基因后,能改善胞内还原力不足的状况,有效提高胞内TCA循环速率。
2.5 基因敲除对细菌的生长影响
以M9培养基为发酵培养基,检测了野生型大肠杆菌MG1655以及经基因敲除所得M06、M07菌株在发酵处理36 h时的生长情况及相应的葡萄糖消耗量,结果分别如图6和图7所示。结合图6和图7可见,上述3种菌株在发酵处理36 h时,M9培养基中的葡萄糖逐渐消耗殆尽,在此期间,三者按生长速率和葡萄糖消耗速率从大到小的排序为:野生型大肠杆菌MG1655、M07、M06菌株。可见相关基因的敲除对细菌的生长造成了一定的影响,相比于野生型大肠杆菌MG1655,菌株M06和M07菌株的生长速率均有所下降,同时,由于pfkA基因的敲除使得M07菌株胞内氧化还原更加平衡,所以其生长速率较M06菌株相应值更大。总的来说,基于野生型大肠杆菌MG1655,进行相关基因敲除后对所得M07菌株的生长影响较小,后者具有成为生产L-苹果酸的工程菌株的潜力。

图6 菌株的生长曲线

图7 菌株的葡萄糖消耗曲线
3 结论
(1)以pTargetF质粒做模板,借助引物通过全质粒PCR扩增,成功构建了基因敲除载体pTargetF-sgRNA。以大肠杆菌MG1655基因组为模板,借助引物扩增相关基因的上、下游同源臂,再以相关基因上、下游同源臂做模板,借助引物通过SOE-PCR进行扩增,成功制得相应基因上、下游同源臂的融合片段。
(2)在敲除载体构建完毕并制得同源臂片段后,利用CRISPR/Cas9基因编辑系统对野生型大肠杆菌MG1655进行基因敲除,依次敲除poxB、pflB、maeB、ldhA、maeA、pta-ackA、pfkA基因后得到菌株M07,经好氧发酵48 h后所得L-苹果酸积累量为9.893 g/L,得率为66%,相比野生型大肠杆菌MG1655相应值明显增加,且基因敲除对该菌株的生长影响不大。
猜你喜欢
工业微生物(2024年1期)2024-02-29 07:36:50
生物加工过程(2023年6期)2023-12-11 03:27:52
睿士(2023年9期)2023-09-20 05:47:07
牡丹江医学院学报(2021年5期)2021-12-05 08:01:51
生物工程学报(2019年1期)2019-01-30 08:19:58
小雪花·初中高分作文(2016年9期)2016-05-14 02:50:08
食品科学(2013年23期)2013-03-11 18:30:10
中国果业信息(2013年12期)2013-01-22 12:16:58
中国烟草学报(2012年2期)2012-04-09 06:44:52
中国医学科学院学报(2012年3期)2012-03-25 13:58:57
