
白藜芦醇的合成
2020-10-26 01:46丁华平顾祁昕蒋程飞张树伟
扬州大学学报(自然科学版) 2020年4期
丁华平, 顾祁昕, 蒋程飞, 张树伟, 袁 宇*
(1. 扬州大学化学化工学院, 江苏 扬州 225002; 2. 江苏长青农化股份有限公司, 江苏 扬州 225200)
白藜芦醇, 即(4-羟苯基)-乙烯基-1, 3-苯二酚(3, 4′, 5-trihydroxy-trans-stilbene), 又名芪三酚[1], 是一种天然抗氧化剂[2-3], 可以降低血液黏稠度, 抑制血小板凝结和血管舒张,保持血液畅通,预防冠心病、缺血性心脏病,对肿瘤也具有一定抑制作用[4-5].目前, 白藜芦醇主要用于护肤品、保健品等领域[6-8].白藜芦醇的生产方法主要分为生物提取和化学合成[9].但由于植物体内白藜芦醇含量少, 生物提取的技术难度较高, 所以化学合成法具有更高的生产实用性.已报道的白藜芦醇的化学合成法主要有Wittig法、Perkin法和Heck法.Wittig法[10]主要是从3, 5-二羟基苯甲酸为起始原料制成相应的Wittig盐, 然后再和对应醛的衍生物进行缩合, 最终脱保护得到产物,但是该方法原子经济性不高,而且立体选择性较差,存在顺反式产物; Perkin法[11]是用3, 5-二羟基苯甲醛作为起始原料,然后跟酸酐进行缩合, 再脱羧、去保护合成白藜芦醇,所得均为反式产物, 但在Perkin法中脱羧步骤的反应条件较为苛刻,反应温度高达260 ℃; Heck法[12]是利用3, 5-二乙酰氧基苯乙烯与对乙酰氧基碘苯发生Heck反应, 然后水解脱保护即可, 该法的反应条件较为温和, 但原料对乙酰氧基碘苯非常昂贵,不利于工业化生产.以上方法均由2个带有酚羟基保护基的化合物经缩合反应再脱保护制得白藜芦醇, 虽然工业处理并不复杂,但是最后脱保护步骤须脱去3个分子的保护基, 会产生超过原料质量10倍以上的工业污水,污染环境的同时增加了生产成本.
本文拟在已有白藜芦醇合成路线基础上进行方法改进, 减少酚羟基的保护以及脱保护产生的工业废水,以对甲氧基肉桂醛为起始原料,在碱性条件下和丙酮发生羟醛缩合反应, 然后脱去甲基,上苄基保护基, 再环合、水解、脱羧、脱氢和脱保护得到白藜芦醇.该方法可大大减小生产上的安全风险和“三废”的生成,有利于实现工业化生产.
1 实验部分
1.1 主要仪器与试剂
6460LCMS型液质联用仪(安捷伦科技有限公司, 德国)、400 MHz DD2型核磁共振仪(安捷伦科技有限公司, 德国)、低温冷却液循环泵(郑州长城科工贸有限公司, 中国)、鼓风式干燥箱(上海欧迈科学仪器有限公司,中国).对甲氧基肉桂醛(萨恩化学技术有限公司)、丙二酸二乙酯(萨恩化学技术有限公司)、钯的质量分数为5%的钯碳(国药集团)、乙烯(南京特种气有限公司)、其他溶剂均为国药试剂,分析纯.
1.2 白藜芦醇的合成
为减少酚羟基的保护,减少脱保护过程中工业废水的产生, 本文设计如图1所示的合成路线: 以对甲氧基肉桂醛为起始原料,在碱性条件下和丙酮发生羟醛缩合反应,然后脱去甲基,上苄基保护基,再环合、水解、脱羧、脱氢和脱保护得到白藜芦醇.该路线最大的特点在于通过自身反应构建白藜芦醇的其中2个酚羟基,符合原子经济性合成路线的要求.在合成路线中使用苄基作为唯一的保护基团,最后通过钯碳催化使氧化脱氢反应构建芳环与氢气还原脱除苄基在一锅法条件下同时完成.构建芳环时氧化脱除的氢气可作为脱除苄基的原料,无须在体系中设置放氢装置,也无须加入氢气,大大减小了生产安全风险和“三废”的产生.
1.2.1 6-(4-甲氧基苯基)-3, 5-己二烯-2-酮(化合物2)的合成
将1.62 g(0.01 mol)对甲氧基肉桂醛、15 mL丙酮、20%氢氧化钠溶液5 mL置于50 mL单口烧瓶中, 充分溶解, 升温至40 ℃, 搅拌反应2 h, 当薄层层析法(TLC)(展开剂为V(乙酸乙酯(EA))∶V(石油醚(PE))=1∶4)检测到原料完全消失时停止反应.温度降至室温后, 缓慢加入15 mL水, 逐渐有黄色固体析出, 震荡至固体完全析出, 抽滤并水洗3~4次, 滤饼真空干燥12 h, 得到化合物2, 收率为96%.1H NMR (400 MHz, CDCl3)δ: 7.41(d,J=8.6 Hz, 2H), 7.24 (d,J=3.1 Hz, 1H), 6.93~6.85 (m, 3H), 6.80~6.69 (m, 1H), 6.21 (d,J=15.5 Hz, 1H), 3.82 (s, 3H), 2.30 (s, 3H).
1.2.2 6-(4-羟基苯基)-3, 5-己二烯-2-酮(化合物3)的合成
将2.02 g(0.01 mol)化合物2, 2.3 g吡啶盐酸盐, 6 mL乙酸酐, 置于装有冷凝管的25 mL三口烧瓶中, 氮气保护下反应2 h.TLC(展开剂为V(EA)∶V(PE)=1∶1)结果显示原料基本消失, 停止反应.待温度降至室温后, 向体系中加入10 mL质量百分数为20%的氢氧化钠溶液, 加热搅拌反应20 min, 并用二氯甲烷洗涤5~6次, 除去杂质.最后在水相中加入稀盐酸, 调至弱酸性, 即有大量黄色固体析出, 抽滤出的滤饼真空干燥12 h, 得到化合物3, 收率68%.1H NMR (400 MHz, DMSO-d6)δ: 9.83 (s, 1H), 7.42~7.29 (m, 3H), 7.00 (d,J=15.5 Hz, 1H), 6.89 (d,J=10.6 Hz, 1H), 6.77~6.72 (m, 2H), 6.13 (d,J=15.5 Hz, 1H), 2.21 (s, 3H).
1.2.3 6-(4-苯基苄氧基)-3, 5-己二烯-2-酮(化合物4)的合成
将1.88 g(0.01 mol)化合物3、2.04 g溴化苄和2.07 g碳酸钾置于50 mL的单口圆底烧瓶, 加入24 mL四氢呋喃溶液, 升温至70 ℃, 加热回流3 h, TLC(展开剂为V(EA)∶V(PE)=1∶1)结果显示原料完全消失时, 停止反应.待温度降至室温, 向体系中加入20 mL水淬灭反应, 接着用100 mL乙酸乙酯萃取5~6次, 向油相中加入无水硫酸钠固体静置干燥6 h, 过滤后减压蒸馏得到黄色固体, 即化合物4, 收率93%.1H NMR (400 MHz, DMSO-d6)δ: 7.51 (d,J=8.7 Hz, 2H), 7.46~7.27 (m, 6H), 7.08~6.93 (m, 4H), 6.16 (d,J=15.5 Hz, 1H), 5.11 (s, 2H), 2.22 (s, 3H).
1.2.4 2-(4-苄氧基苯乙烯基)-4, 6-二氧代环己烷羧酸乙酯(化合物5)的合成
将0.299 g(0.013 mol)钠块、6 mL无水乙醇置于50 mL的三口烧瓶中, 充分搅拌混合均匀, 回流, 待气体不再放出, 加入1.76 g(0.011 mol)丙二酸二乙酯, 室温下搅拌15 min, 待反应体系变成白色乳状液体, 最后加入用6 mL无水乙醇溶液溶解的2.78 g(0.01 mol)化合物4, 80 ℃下回流3 h, TLC(展开剂为V(乙酸)∶V(EA)=1∶30)结果显示原料完全消失, 停止反应.待温度降至室温, 减压蒸馏,得到的固体用体积比为1∶ 3的乙酸乙酯/石油醚混合液洗涤3~4次后真空干燥8 h,得到化合物5, 收率92%.1H NMR (400 MHz, CDCl3)δ: 7.47~7.15 (m, 7H), 6.95~6.84 (m, 2H), 6.43 (d,J=15.8 Hz, 1H), 6.23 (d,J=15.8 Hz, 1H), 5.86 (dd,J=15.8, 6.6 Hz, 1H), 5.06 (d,J=56.8 Hz, 2H), 4.32~4.08 (m, 2H), 3.87 (dd,J=8.4, 3.3 Hz, 1H), 3.16 (d,J=12.0 Hz,1 H), 2.82~2.32 (m, 3H), 1.26 (t, 3H).
1.2.5 2-(4-苄氧基苯乙烯基)-4, 6-二氧代环己烷羧酸(化合物6)的合成
将3.92 g(0.01 mol)化合物5、0.44 g氢氧化钠固体置于50 mL圆底烧瓶中, 加入水(17 mL)和甲醇(4 mL)的混合溶剂, 搅拌均匀, 100 ℃下回流反应3 h, TLC(展开剂为V(乙酸)∶V(EA)=1∶30)跟踪反应, 直至原料完全消失, 停止反应.待温度降至室温, 减压蒸馏除去多余溶剂, 收集固体产品, 即化合物6, 收率约95%.1H NMR (400 MHz, CDCl3)δ: 7.49~7.13 (m, 7H), 6.98~6.82 (m, 2H), 6.43 (d,J=15.8 Hz, 1H), 6.23 (d,J=15.8 Hz, 1H), 5.06 (d,J=16.8 Hz, 2H), 4.42~3.91 (m, 2H), 3.59 (d,J=11.1 Hz, 1H), 2.80~2.38 (m, 3H).
1.2.6 5-(4-苄基氧基苯乙烯基)环己-1, 3-二酮(化合物7)的合成
将3.64 g(0.01 mol)化合物6置于50 mL三口烧瓶中, 加入18 mL水, 缓慢升温至60 ℃, 充分搅拌使原料完全溶解, 向体系中缓慢滴加3 mL浓度为12 mol·L-1的浓盐酸, 升温至100 ℃, TLC(展开剂为V(甲醇)∶V(EA)=1∶20)跟踪反应, 直至原料完全消失, 停止反应.待温度降至室温, 体系有淡黄色固体析出, 抽滤, 得到淡黄色固体.石油醚洗涤2~3次, 除去表面的油状物, 再用V(EA)∶V(PE)=1∶3混合溶剂重结晶除去杂质, 真空干燥, 得到化合物7, 收率91%.用HPLC归一化法检测产物, 纯度达97.5%以上.1H NMR (400 MHz, DMSO-d6)δ: 7.49~7.20 (m, 7H), 6.93 (d,J=8.7 Hz, 2H), 6.36 (d,J=16.0 Hz, 1H), 6.11 (dd,J=16.0, 6.8 Hz, 1H), 5.20 (s, 1H), 5.06 (s, 2H), 2.84 (dd,J=9.3, 6.4 Hz, 1H), 2.23 (dd,J=45.7, 10.7 Hz, 4H).
1.2.7 白藜芦醇(化合物8)的合成
将3.2 g(0.01 mol)化合物7、4.24 g钯碳(钯含量为0.212 g, 0.002 mol)和17 mL乙腈置于100 mL内胆高压反应釜, 通入氮气排除体系中多余的空气, 保持反应釜内的压力约0.3 MPa, 反应温度80 ℃, 搅拌24 h.反应结束后, 抽滤回收钯碳,减压蒸馏除去溶剂, 得到白色固体即为目标产物8, 产品收率82%.1H NMR (400 MHz, 丙酮-d6)δ: 8.30 (d,J=107.0 Hz, 3H), 7.44~7.36 (m, 2H), 7.02 (dd,J=12.4, 7.6 Hz, 1H), 6.94~6.78 (m, 3H), 6.53 (d,J=2.1 Hz, 2H), 6.26 (t,J=2.2 Hz, 1H).
2 结果与讨论
该路线的关键是最后通过钯碳催化使氧化脱氢反应构建芳环与氢气还原脱除苄基在一锅法条件下同时完成, 钯碳在这一过程中起到2种作用: 反应物在钯碳的作用下脱去1分子氢气,然后钯碳上所吸附的氢分子再进攻苄基,脱去保护基.由于该反应在反应过程中存在递进关系,所以钯碳的用量尤为重要.在本文实验条件下, 钯碳用量对反应收率的影响见表1.如表1所示,当钯碳的物质的量是化合物7的0.05倍时, 产品收率仅18%, 这是因为过少的钯碳只能催化其进行脱氢还原反应, 脱保护反应不完全, 产物中存在大量苄基保护的副产物;当提高钯碳的物质的量至化合物7的0.2倍时, 反应效果较好,产品收率达82%; 继续增加钯碳的用量对反应产率没有明显影响,因此控制化合物7和钯碳的物质的量比为1∶0.2.
在这一步中,体系的压强也会影响收率,当控制化合物7与催化剂的物质的量比为1∶0.2, 乙腈为反应溶剂,反应温度为80 ℃时,考察压强对反应产率的影响,结果见表2.由表2中可知,体系压强对反应收率影响较大.当压强为0.3 MPa时,收率可达82%,这是因为脱保护反应类似于加氢反应,需要较高的压强,否则反应平衡会朝着反方向进行;但是当压强继续增加至0.5 MPa时,压强的增大阻碍了脱氢反应的进行,致使收率减低: 因此反应适合的压强为0.3 MPa.
针对化合物7溶解度较差的问题, 本文对溶剂的用量进行了考察. 在压强为0.3 MPa,温度为80 ℃, 化合物7的物质的量为0.01 mol的实验条件下, 不同用量的溶剂对应的收率见表3.实验结果表明溶剂乙腈的用量对反应收率有较大影响,由于原料在溶剂中的溶解度较差,适当增加溶剂的用量,可明显提高收率,当溶剂体积为17 mL时, 反应收率可达82%.

表1 钯碳用量对反应收率的影响
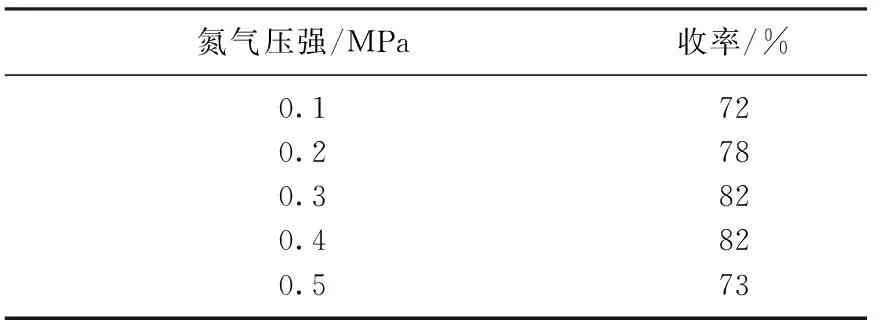
表2 氮气压力对反应收率的影响

表3 溶剂的用量对收率的影响
3 结论
本合成路线主要是选用对甲氧基肉桂醛为起始原料,然后选用苄基作保护基合成白藜芦醇, 经过羟醛缩合反应、去保护基、上保护基、环合、水解、脱羧、脱氢和脱保护制得白藜芦醇.总反应收率达40%.本文方法条件比较温和、操作便捷、对环境友好、每步反应收率都较高,相较于传统路线,有较高的工业生产潜力.
猜你喜欢
工业催化(2022年9期)2022-10-17
云南化工(2022年2期)2022-03-18
花生学报(2021年1期)2021-08-30
天津医科大学学报(2021年4期)2021-08-21
农药科学与管理(2021年2期)2021-03-16
中国化工贸易·上旬刊(2019年3期)2019-09-10
健康之家(2016年12期)2017-01-05
中国科技纵横(2016年13期)2016-08-22
湖南大学学报·自然科学版(2014年3期)2014-12-30
