
HPLC法同时测定大柴胡颗粒中芍药苷和黄芩苷的含量
2020-10-15 10:44袁文静
中国民族民间医药 2020年17期
袁文静
河南省周口市食品药品检验所,河南 周口 466000
大柴胡颗粒源于汉代张仲景《金匮要略》中记载的“大柴胡汤”,该方主要有柴胡、大黄、枳实(炒)、黄芩、芍药、半夏(姜)、大枣、生姜8味药组成[1-3],具有和解少阳、内泻热结的功效,现代常用于治疗因少阳不和、肝胆湿热所致的口苦、舌红苔黄腻、大便秘结,临床应用较为广泛[4-5]。目前关于大柴胡颗粒有效成分的含量测定报道少较,药典中也尚未有质量标准的记载,因此建立完善、系统的质量标准,对大柴胡颗粒安全有效的应用于临床有重要意义。黄芩苷和芍药苷分别是黄芩和白芍的主要有效成分[6-7],本研究采用快速、高效的HPLC技术同时测定大柴胡颗粒中芍药苷和黄芩苷的含量,为建立大柴胡颗粒质量标准提供科学依据。
1 仪器与试药
1.1 仪器 Agilent-1260高效液相色谱仪,配有G1311C 1260 Quat Pump VL 四元低压泵,G1316A 1260 Tcc 恒温箱,G1329B 1260 Als自动进样器,G1314F 1260 VWD 型紫外检测器,Rev.B.04.03[52]Agilent Chem-station数据处理系统(安捷伦科技有限公司);AE-100电子天平(德国梅特勒公司)
芍药苷对照品,批号为110736-201741,纯度为95.70%,黄芩苷对照品,批号为110781-201717,纯度为96.9%,均来源于中国食品药品检定研究院;大柴胡颗粒(精华制药集团股份有限公司,批号分别为 100103、110801、110802);蒸馏水(可帮化工有限公司);甲醇、乙腈为色谱纯(科密欧公司);磷酸为分析纯(西域化学试剂公司)。
2 方法与结果
2.1 色谱条件与系统适用性试验 采用Kromasil 100-5C18柱(250 mm×4.6 mm,5 μm)进行检测;流动相为乙腈(A)-0.05%磷酸水(B),采用梯度洗脱的方法,程序为0~15 min(5%~20% A),15~28 min(20%~25%A),28~37 min(25%~25% A),37~50 min(25%~55% A),柱温为30 ℃;流速为0.8 mL/min;检测波长:采用分段变波长模式,0~20 min,检测波长为230 nm;20~30 min,检测波长为280 nm;进样量为10 μL。理论塔板数要求:按黄芩苷计算不得低于4000,按芍药苷计算不得低于3000。
2.2 对照品及供试品溶液的制备
2.2.1 对照品溶液的制备 精密称取芍药苷对照品20.08 mg,置于100 mL的容量瓶中,然后加甲醇定容至刻度,超声溶解混匀,得芍药苷对照品的储备液;精密称取黄芩苷对照品45.25 mg,置于50 mL的容量瓶中,加甲醇定容至刻度,超声溶解混匀,得黄芩苷对照品储备液。分别精密量取芍药苷、黄芩苷对照品储备液适量,置于同一容量瓶中,加甲醇稀释至刻度,配置成每1 mL含芍药苷0.1 mg、含黄芩苷0.4525 mg的混合对照品溶液。
2.2.2 供试品溶液的制备 取大柴胡颗粒适量,研细,精密称定1.0 g,置于25 mL的容量瓶中,加入10 mL浓度为75%甲醇溶液进行溶解,然后超声处理15 min,此操作重复处理2次,待溶液放冷后,再加入75%甲醇定容至刻度,摇匀,检测前过0.45 μm的微孔滤膜,即得到供试品溶液。
2.2.3 阴性对照溶液的制备 依照生产厂家大柴胡颗粒原配方比例分别称取缺白芍、缺黄芩以外的其余药材,然后按照厂家的生产工艺分别制成缺黄芩、缺白芍的阴性样品颗粒,随后按照“2.2.2”项下步骤进行操作,制成缺白芍、缺黄芩的阴性对照溶液。
2.3 方法学考察
2.3.1 阴性干扰实验 分别取黄芩苷和芍药苷的混合对照品溶液、缺白芍阴性对照溶液、缺黄芩的阴性对照溶液以及供试品溶液,按照设定“2.1”项下的色谱条件依次进样分析。结果在阴性对照品溶液黄芩苷和芍药苷色谱峰相应位置处无干扰峰出现(如图1所示),表明阴性对照无干扰,该方法专属性良好。
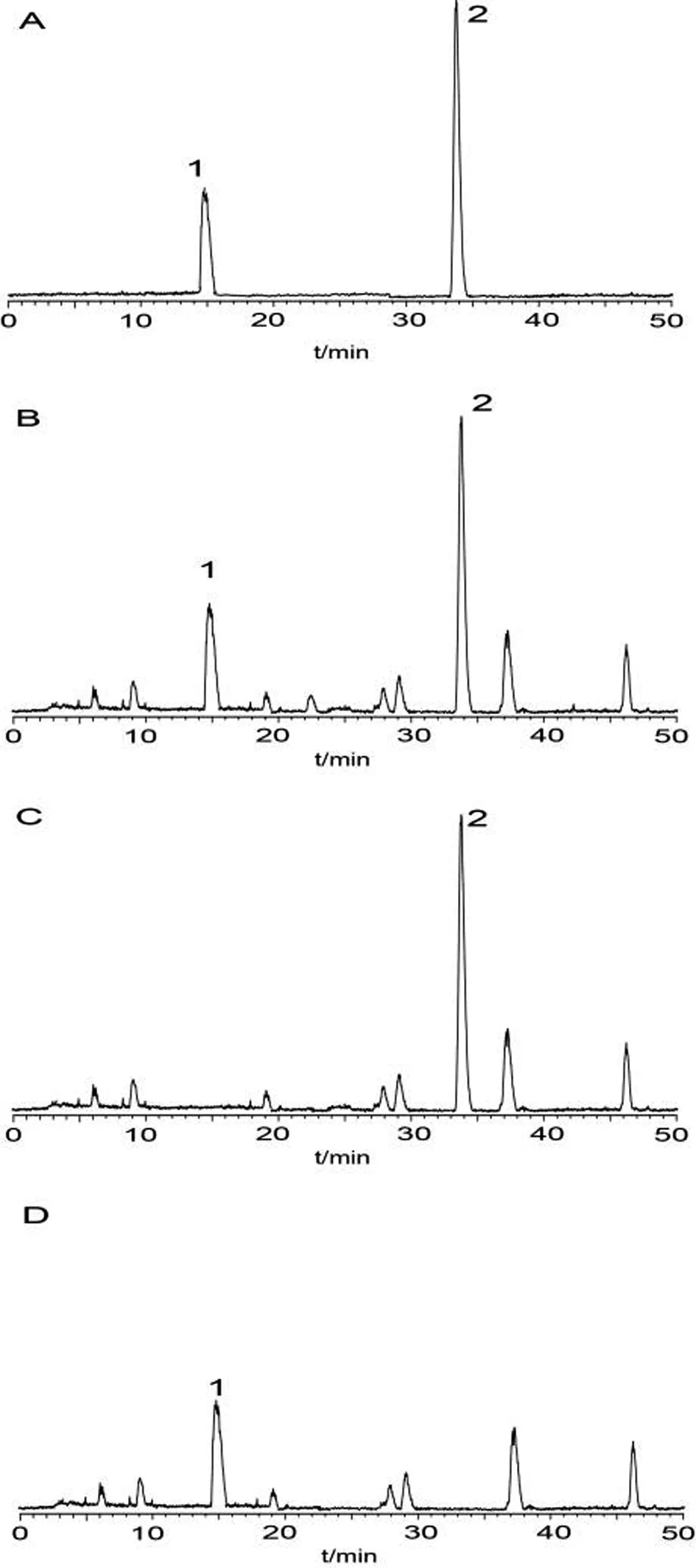
1.芍药苷 2.黄芩苷
2.3.2 线性关系考察 精密吸取“2.2.1”项下芍药苷对照品溶液1、2、5、10、15、20μL,得6份样品,按照“2.1”项下的色谱条件将6份样品依次进样分析并记录相应的峰面积。以进样量(μg)为横坐标,峰面积(A)为纵坐标绘制标准曲线,以最小二乘法求得芍药苷回归方程为:Y=689.72X+15.214(r=0.9999),表明芍药苷进样量在 0.1~2.0 μg范围内线性关系良好。
精密吸取“2.2.1”项下黄芩苷对照品溶液1、2、5、10、15、20 μL,得6份样品,按照“2.1”项下的色谱条件将6份样品依次进样分析并记录相应的峰面积。以进样量(μg)为横坐标,峰面积(A)为纵坐标绘制标准曲线,以最小二乘法求得黄芩苷回归方程为Y=1275.8X+45.213(r=0.9999),表明黄芩苷进样量在 0.4525~9.05 μg范围内线性关系良好。
2.3.3 精密度试验 精密吸取混合对照品适量,按照“2.1”项下色谱条件对同一样品重复进样分析6次,计算芍药苷、黄芩苷色谱峰峰面积及相对应的标准偏差(RSD)值。得芍药苷RSD值为0.87%,黄芪苷的RSD值为1.21%,试验结果表明该仪器精密度良好,可以满足进一步的试验要求。
2.3.4 重复性试验 取相同批号的大柴胡颗粒(批号100103)适量,按照“2.2.2”项下的步骤进行操作,平行制备6份供试品溶液。取制得的6份供试品溶液,分别按照“2.1”项下色谱条件进行进样分析,随后分别记录芍药苷、黄芪苷的色谱峰峰面积,计算其RSD值。得芍药苷RSD值为1.25%,黄芪苷的RSD值为0.98%,试验结果表明该方法重复性良好。
2.3.5 稳定性试验 取同一批号的大柴胡颗粒(批号100103)适量,按照“2.2.2”项下操作制备供试品溶液。取同一供试品溶液适量,分别在置于室温下0、2、4、8、12、24h后,按照“2.1”项下色谱条件进行进样分析,随后分别记录芍药苷、黄芪苷的色欧风峰面积,计算其RSD值。得芍药苷RSD值为1.63%,黄芪苷的RSD值为1.21%,试验结果表明样品24 h内稳定性良好。
2.3.6 加样回收率试验 取同一批号的大柴胡颗粒(批号100103)适量,研细,平行称取6份,每份样品约0.5 g,随机分为3组,每组2份,置于带塞锥形瓶中,3组样品分别加入低、中、高三个浓度的混合对照品(芍药苷2.0053 mg/mL,黄芪苷8.2316 mg/mL)0.8 mL、1.0 mL、1.2 mL,然后按照“2.2.2”项下操作步骤制备供试品溶液,取供试品溶液10 μL,按照“2.1”项下设定的色谱条件进行进样分析,根据加入量和测得量计算加样回收率,结果见表1。
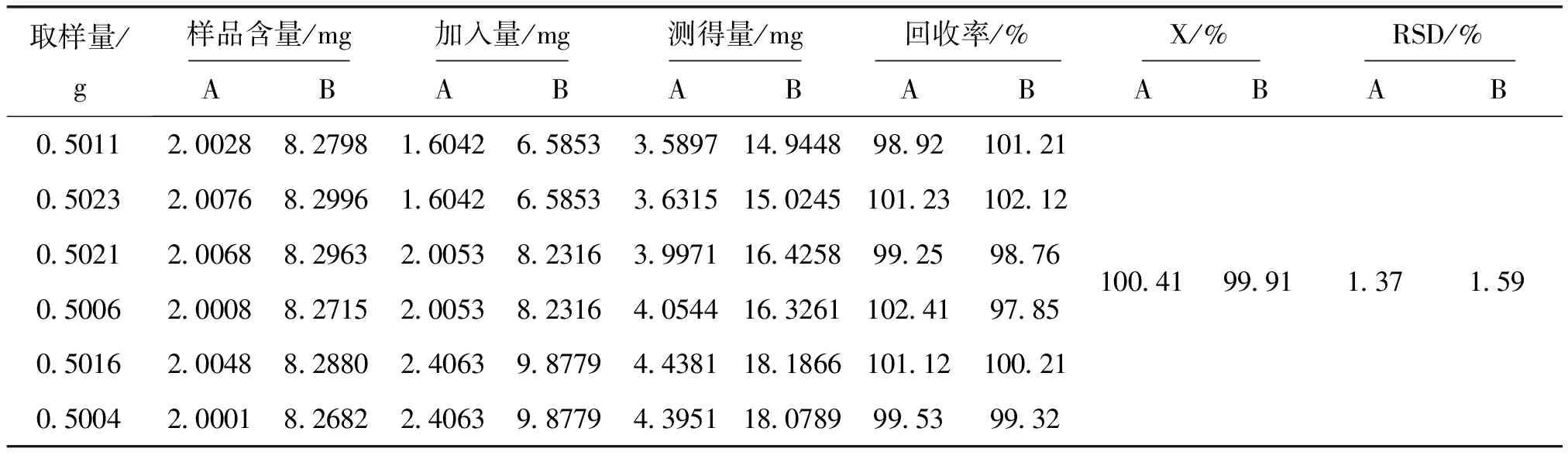
表1 加样回收试验结果 (n=6)
2.3.7 样品含量测定 分别称取3个批次(100103、110801、110802)的大柴胡颗粒适量,按照“2.2.2”项下操作步骤制备得到供试品溶液。分别取每个批次的取供试品溶液10μL按照“2.1”项下设定的色谱条件进行进样分析,得各个批次样品中芍药苷、黄芩苷的含量,结果见表2。
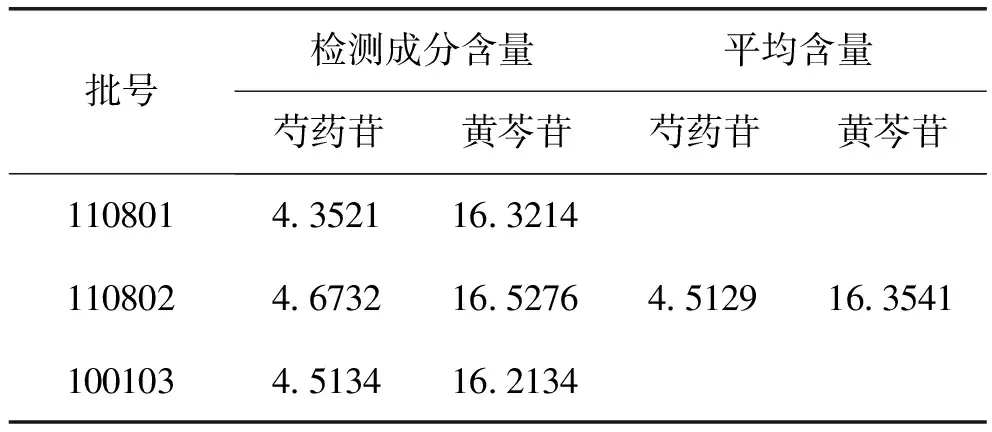
表2 大柴胡颗粒中芍药苷、黄芩苷的含量测定 (mg/g)
3 讨论
3.1 指标性成分的选择 黄芩味苦、性寒,有清热燥湿、泻火解毒的功效。药效学研究表明黄芩中主要的药效成分为黄酮类化合物黄芩苷,同时《中国药典》中规定黄芩苷为黄芩饮片质量控制的检测指标,现代药理研究表明黄芩苷具有显著的生物活性,用于临床有保肝利胆、抗菌、抗炎、抗变态及解痉的药理作用[8-9]。白芍味苦、酸,性微寒,具有养血敛阴、柔肝止痛的功效,白芍的主要药效成分为芍药苷,也是《中国药典》中白芍饮片的质量控制指标,同时现代药理研究表明芍药苷具有抗炎、解热、解痉、调节免疫的作用[10-11]。因此,本研究选择黄芩苷和芍药苷为检测指标。
3.2 流动相的选择 根据有关报道[12-15],本研究曾选择甲醇-水、甲醇-0.1磷酸、乙腈-0.1%冰醋酸、乙腈-0.05磷酸作为流动相,试验结果发现乙腈较甲醇的基线更平稳,黄芩苷在酸性条件下较纯水的峰形更尖锐,灵敏度更高,可能原因为黄芩苷中含有酚羟基,有弱酸性,在流动相中加入少量酸,可有效抑制黄芩苷解离。另外考虑到色谱柱对酸的耐受性,最终选择乙腈-0.05%磷酸作为流动相。
3.3 供试品溶液制备方法的选择 在提取剂选择方面,以色谱峰的分离效果和吸收强度为考察的指标,选取了水、95%乙醇、甲醇为提取溶媒,结果表明甲醇提取液色谱峰的数目以及分离度明显优于其他的提取液,因此最终选择甲醇作为提取溶剂。在提取方式方面,曾尝试回流提取、超声提取、索式提取,结果表明3种方式的提取效率并无太大差别,由于超声提取更加的便捷、节省时间,因此最终选择甲醇为溶媒,进行超声提取。
4 小结
本研究建立HPLC法同时测定大柴胡颗粒中黄芩苷和芍药苷的含量的分析方法,并经过方法学验证,该方法简便、快速,结果可靠,重现性好,可作为大柴胡颗粒中芍药苷、黄芩苷含量测定的方法,为大柴胡颗粒的质量控制提供可参考的检测方法。
猜你喜欢
基层中医药(2022年7期)2022-11-17
今日农业(2022年13期)2022-09-15
山西大同大学学报(自然科学版)(2022年4期)2022-08-29
上海文化(文化研究)(2021年6期)2022-01-07
世界科学技术-中医药现代化(2021年9期)2021-12-31
中国合理用药探索(2021年9期)2021-11-03
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2021年1期)2021-01-26
今日农业(2020年16期)2020-12-14
中成药(2019年12期)2020-01-04
