
猪流行性腹泻病毒流行株S全基因的遗传进化与重组分析
2020-10-14 11:25崔建涛韩昊莹党家刚田润博陈红英郑兰兰杨明凡
中国兽医学报 2020年9期
崔建涛,韩昊莹,党家刚,赵 宇,田润博,陈红英.3*,郑兰兰,杨明凡
( 1.河南农业大学 牧医工程学院,河南 郑州 450002;2.泌阳县农业科学研究所,河南 泌阳463700;3.郑州市猪重大疫病防控重点实验室,河南 郑州 450002)
猪流行性腹泻(porcine epidemic diarrhea,PED)是由猪流行性腹泻病毒(porcine epidemic diarrhea virus,PEDV)引起的一种急性、高度传染性和严重肠道疾病,引起猪呕吐、急性腹泻和脱水以及哺乳仔猪的高死亡率[1-3]。2010年以前,PED在我国各地呈现零星散发,猪群的发病率和致死率很低,2010年底,我国大面积暴发PED疫情,导致猪群高发病率及高死亡率,尤其感染仔猪的致死率高达100%,造成巨大的经济损失[4-6]。2013年,PED在美国流行[7]。到目前为止,PED在全球暴发,给全球养猪业带来巨大经济损失。
PEDV的S基因可以编码位于病毒粒子表面的纤突蛋白,在促进病毒粒子与机体细胞膜融合过程中发挥重要生物学作用[8]。S基因是PEDV重要的毒力基因,由于碱基突变、缺失或插入,不同毒株之间S基因差异较大[9]。众多研究证实,S基因是序列分析的典型基因,在研究PEDV流行性株遗传变异中发挥重要作用。
本研究对2016—2018年间,从河南和山西等地多个规模化猪场随机采集的25份疑似腹泻发病仔猪的小肠及内容物提取RNA进行RT-PCR检测,对检测为阳性的PEDV样品进行S全基因克隆与序列分析,对当前PEDV流行株进行遗传进化与重组分析,为PED选用合适的候选疫苗,对该疾病的防控提供参考依据。
1 材料与方法
1.1 病料样品2016年3月至2018年4月间,从河南和山西等地多个猪场随机采集的25份疑似腹泻仔猪的小肠及内容物。
1.2 主要试剂DNA 胶回收试剂盒购自上海生工有限公司;HiScript®Ⅱ1st Strand cDNA Synthesis Kit购自南京诺唯赞生物公司;pMD18-T Vector购自宝生物工程(大连)有限公司;TRIzon Reagent、2×Es TaqMasterMix、DNA Marker购自康为世纪公司;2 kb DNA Ladder购自北京庄盟国际生物基因科技有限公司。
1.3 引物设计根据GenBank中PEDV全基因组序列(登录号:KT860508、KJ399978、KY649107和AF353511),基于M基因高度保守区,设计1对特异性检测引物(表1)。针对S全基因设计2对特异性引物(S1-F/R和S2-F/R),分别扩增S1和S2基因片段。引物由武汉奥科公司进行合成。

表1 PCR扩增引物
1.4 病毒RNA提取及cDNA合成取少量小肠肠道及其内容物或粪便,用PBS液(含青、链霉素)以1∶5的比例混合,充分研磨,反复冻融3次,12 000 r/min离心5 min。取上清液,利用TRlzon提取样品中总RNA,于-80℃保存备用。按照HiScript®Ⅱ1st Strand cDNA Synthesis Kit进行RNA的反转录得到cDNA,于-80℃保存备用。
1.5 PEDV的PCR检测及S全基因的扩增、克隆及测序以1.4中反转录合成的cDNA为模板,利用引物P检-F/R,PCR检测PEDV。利用引物S1-F/R和S2-F/R,对检测为阳性样品cDNA,PCR扩增PEDV S1和S2基因,回收目的基因片段,连接到pMD18-T,送武汉奥科进行测序。
1.6 S全基因序列分析利用DNAStar 5.0软件,将测序获得的PEDV S1和S2基因序列进行拼接,使用MEGA 7.0软件,将本试验的18条PEDV毒株 S全基因序列与30株PEDV参考序列S基因(表2)进行核苷酸以及氨基酸的同源性分析、构建遗传进化树,且用RDP4.39软件对S全基因序列进行重组分析。
2 结果
2.1 PCR检测结果以25份肠道病料样品的cDNA为模板,进行PCR扩增、电泳鉴定,在421 bp处出现特异性条带(图1),与预期扩增片段大小相符。25份肠道病料样品中有18份样品RT-PCR扩增为阳性,阳性率为72%。将回收纯化的PCR扩增产物克隆到pMD18-T载体中进行测序,测序结果显示,所克隆的部分M基因与PEDV参照株(登录号为AF353511、KT860508、KJ399978和KY649107)M基因核苷酸同源性在99%以上,表明RT-PCR扩增具有特异性。
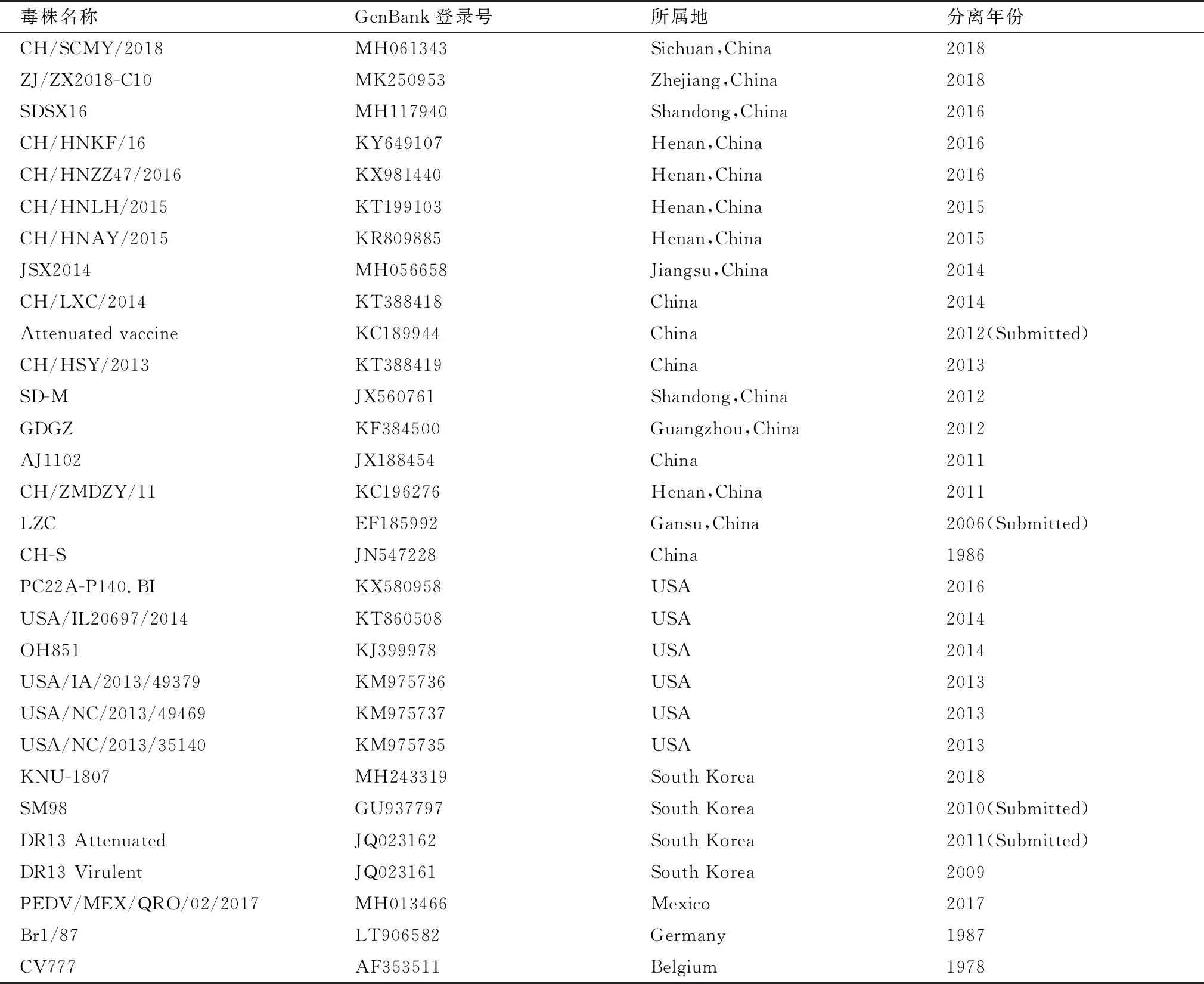
表2 参考毒株序列
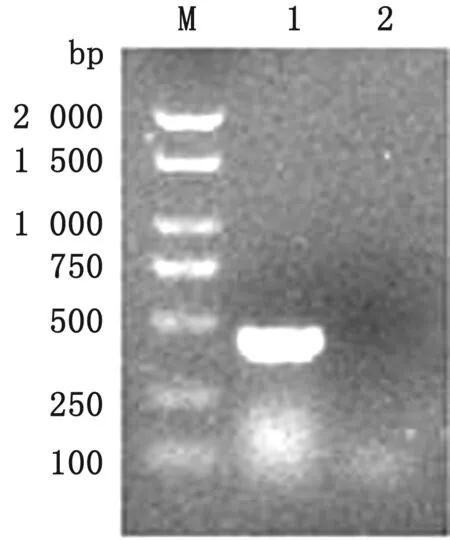
图1 PEDV检测结果 M.DL2000 DNA Marker;1.PEDV 检测结果;2.阴性对照
2.2 S基因扩增及测序结果经PCR扩增及电泳鉴定,S1基因和S2基因分别在约2 400和2 100 bp位置出现1条特异性条带(图2)。测序结果显示,共获得18条PEDV S1和S2序列,通过DNAStar 5.0软件拼接,得到18条完整的PEDV S基因序列,根据样品采集地点分别命名,同时将克隆的S基因序列登录至GenBank,获得GenBank登录号(表3)。
2.3 S基因的同源性分析运用DNAStar 5.0和MEGA 7.0软件对核苷酸的同源性进行分析,结果表明18株PEDV S全基因之间的同源性为96.0%~99.9%,与参考株同源性为92.8%~99.3%。其中与河南往年分离株的同源性为96.2%~99.3%,与经典CV777毒株的核苷酸同源性为93.2%~93.7%,与美国新型毒株USA/IL20697/2014同源性为93.8%~96.0%。本试验克隆的18条PEDVS全基因序列与30条参考毒株相比,存在多处的点突变、碱基插入和缺失。与经典毒株CV777对比,流行株在164~166 nt处有3个碱基(TTG)插入,177~185 nt有9个碱基(GGGTGTCAA)插入,在427~429 nt有3个碱基(AAT)插入,在3 610~3 612 nt 处有个别毒株中有3个碱基(CAT)插入,在487~492 nt有6个碱基(CGTGAT)缺失。推导的S蛋白氨基酸序列比对,发现在氨基酸第55~56位插入2个氨基酸(异亮氨酸和甘氨酸),在59~62位插入4个氨基酸(谷氨酰胺、甘氨酸、缬氨酸和天冬氨酰),在143位插入1个天冬氨酸,在163~164位缺失2个氨基酸(精氨酸和天冬氨酸)。本试验克隆的SX/TY1/2017、SX/TY2/2017、HN/KF/2017、HN/XZ/2017和HN/JY/2018 5株毒株序列相比克隆的其他13株,在3 589~3 591 nt有3个碱基(CAT)缺失。
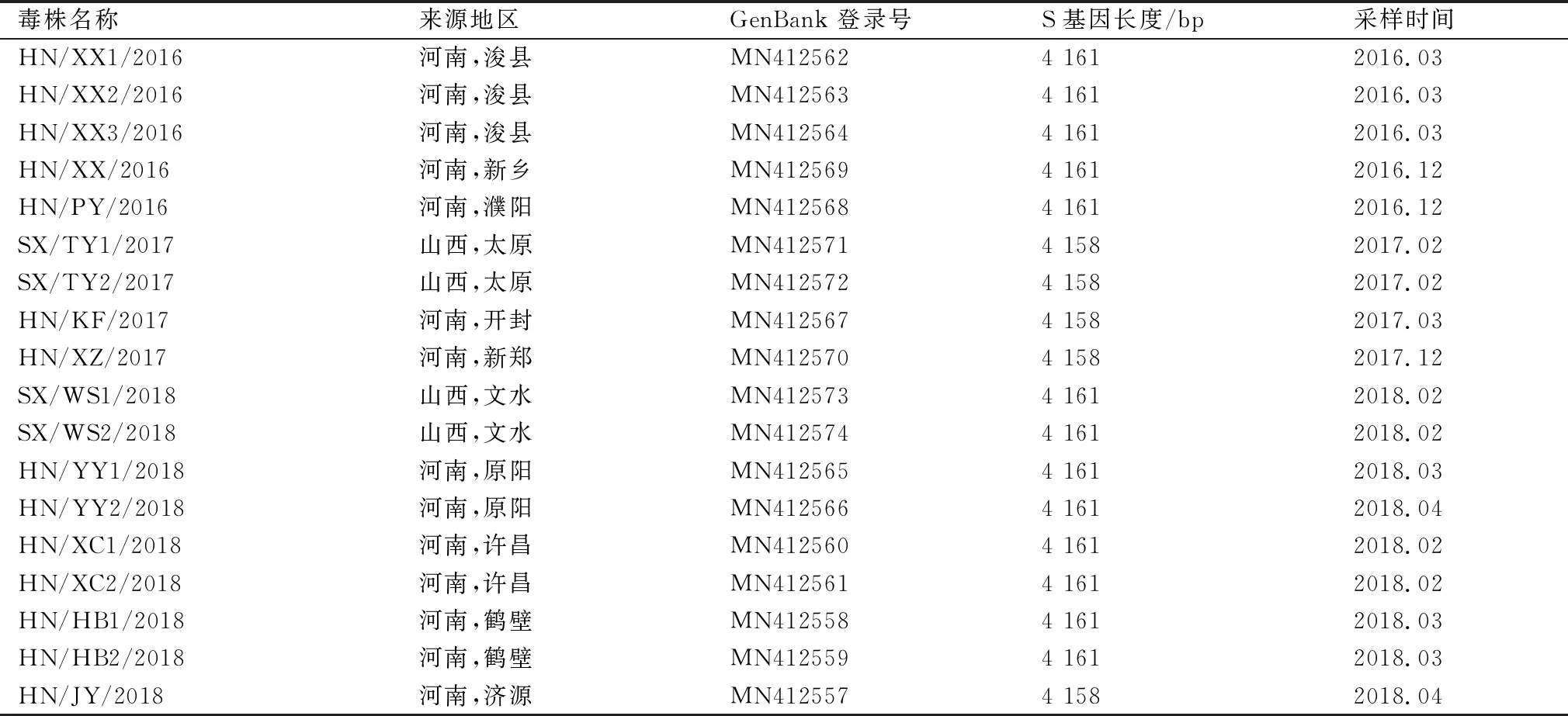
表3 PEDV S基因序列样品信息
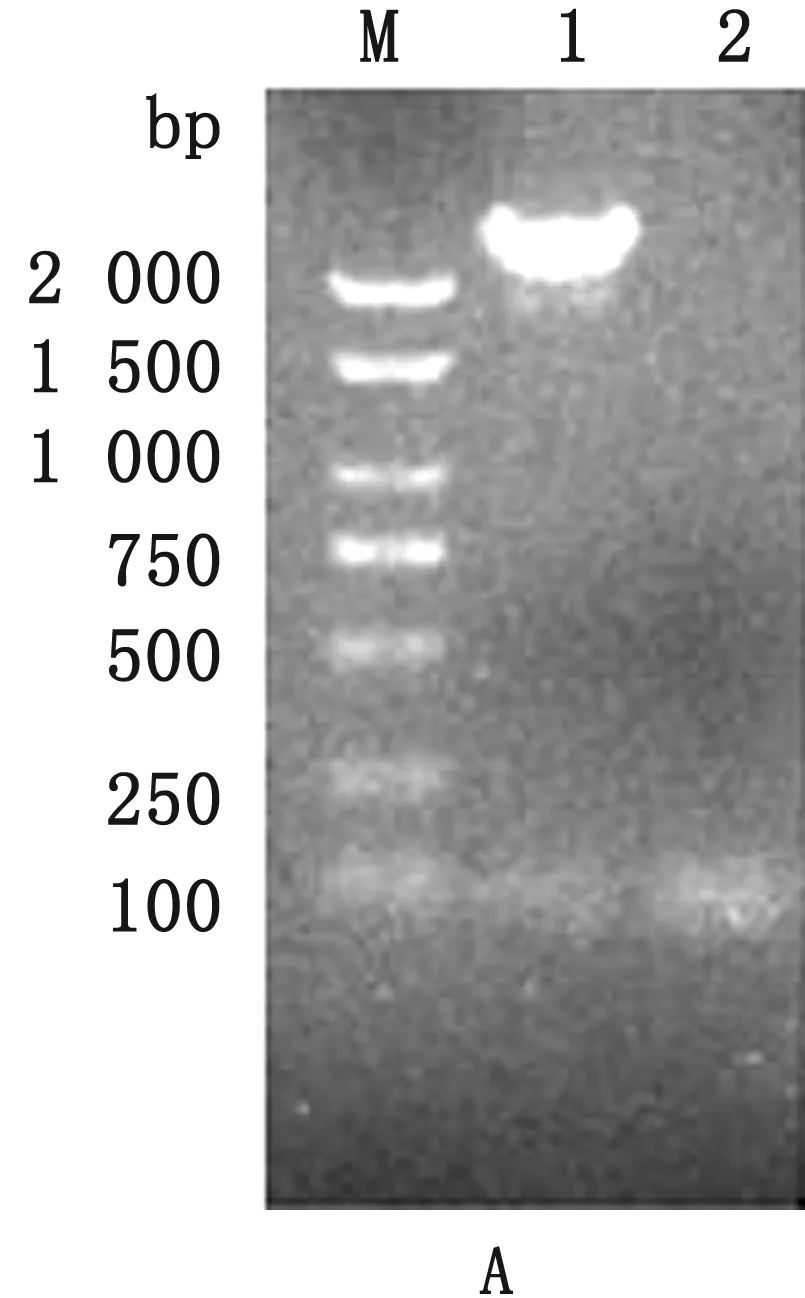
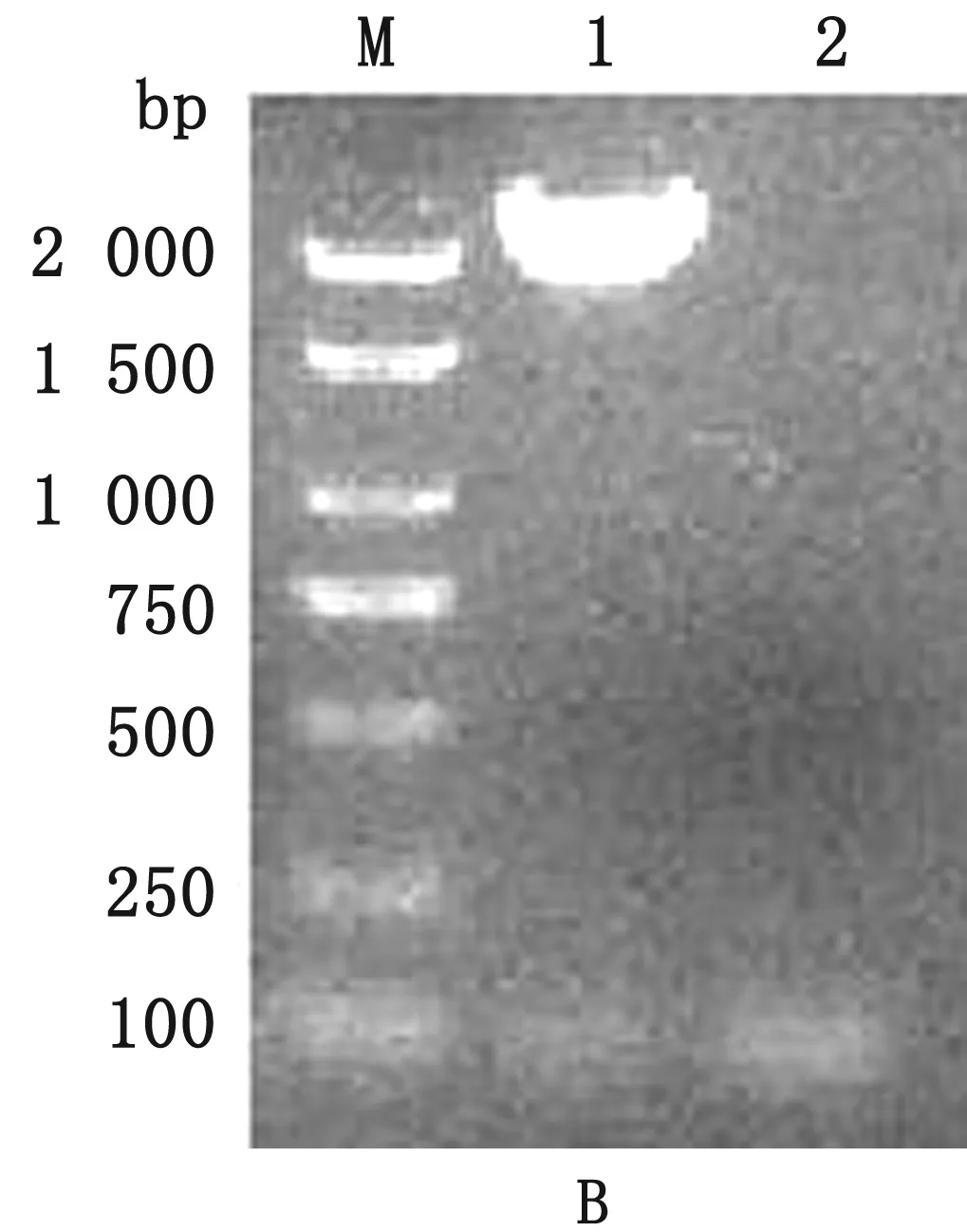
图2 PEDV S1(A)和PEDV S2基因(B)扩增结果 M.DL2000 DNA Marker;A1.S1基因;B1.S2基因;A1,B2.阴性对照
2.4 S基因的遗传进化树通过MEGA 7.0软件对PEDV S全基因序列遗传演化分析,如图3所示,PEDV毒株分为两个群。G1群包括12个毒株,有经典毒株、疫苗株和早些年分离株。G2群包括本试验克隆的18株毒株以及其他参考毒株,主要是2010年以后流行株。本试验克隆的18株毒株处于不同分支,HN/KF/2017、HN/XZ/2017、SX/TY1/2017、HN/JY/2018和SX/TY2/2017与GDGZ、AJ1102,HN/XX/2016、HN/XX3/2016、HN/XX1/2016与CH/LXC/2014以及SX/WS2/2018与CH/HNLH/2015、CH/HNAY/2015、CH/HNKF/16和CH/HSY/2013分别处于一个小的分支,具有较近的亲缘关系。其余克隆毒株独立成一个小分支。
2.5 S基因的重组分析通过RDP4.39软件对PEDV S全基因序列分析,结果显示,克隆的SX/TY1/2017毒株S全基因有1个重组开始位点位于3 291 nt,终止位点在4 132 nt位置(图4),CH/HNZZ47/2016参考毒株和克隆的HN/KF/2017毒株被认为是亲本株。CH/HNZZ47/2016参考毒株是主要亲本株,克隆的HN/KF/2017毒株是次要亲本株。SX/TY1/2017毒株S全基因与CH/HNZZ47/2016毒株和HN-KF-2017毒株的同源性均为97.4%。与经典CV777毒株的核苷酸同源性为93.4%,与美国新型毒株USA/IL20697/2014同源性为95.0%。
3 讨论
自2010年底以来,PED在我国大面积暴发,哺乳仔猪的高发病率和高致死率,给养猪业造成严重危害,同时对我国的经济发展造成巨大损失[10]。有研究发现,临床PEDV、TGEV和猪轮状病毒(PoRV)常常发生混合感染,但是,近几年PEDV是造成猪腹泻甚至死亡的主要病原体,TGEV和PoRV在腹泻猪有一定的检出率,甚至为零检出率[11-13]。2010年,甘振磊等[14]在对腹泻病料进行阳性率检测时发现,PEDV的阳性率(46%)远远高于TGEV(15%)和PoRV(8%),2015年,南文金等[15]对粤北地区采集的腹泻病料进行检测,只检测到PEDV,且阳性率为83.87%,远远高于TGEV和PoRV。以上研究结果表明,PEDV依然是导致腹泻的主要病原体。通过对PEDV的基因组结果以及遗传进化分析的研究发现,导致PEDV在我国暴发和流行的原因是由于宿主免疫选择的变化和病毒自身进化的需要,而PEDV S基因在病毒感染和流行过程中相比较于其他保守基因(比如M、N基因)更易发生变异,这对PED的防制带来了更大的困难,因此,PEDV S基因常常是PEDV各流行毒株间遗传进化分析的靶基因。
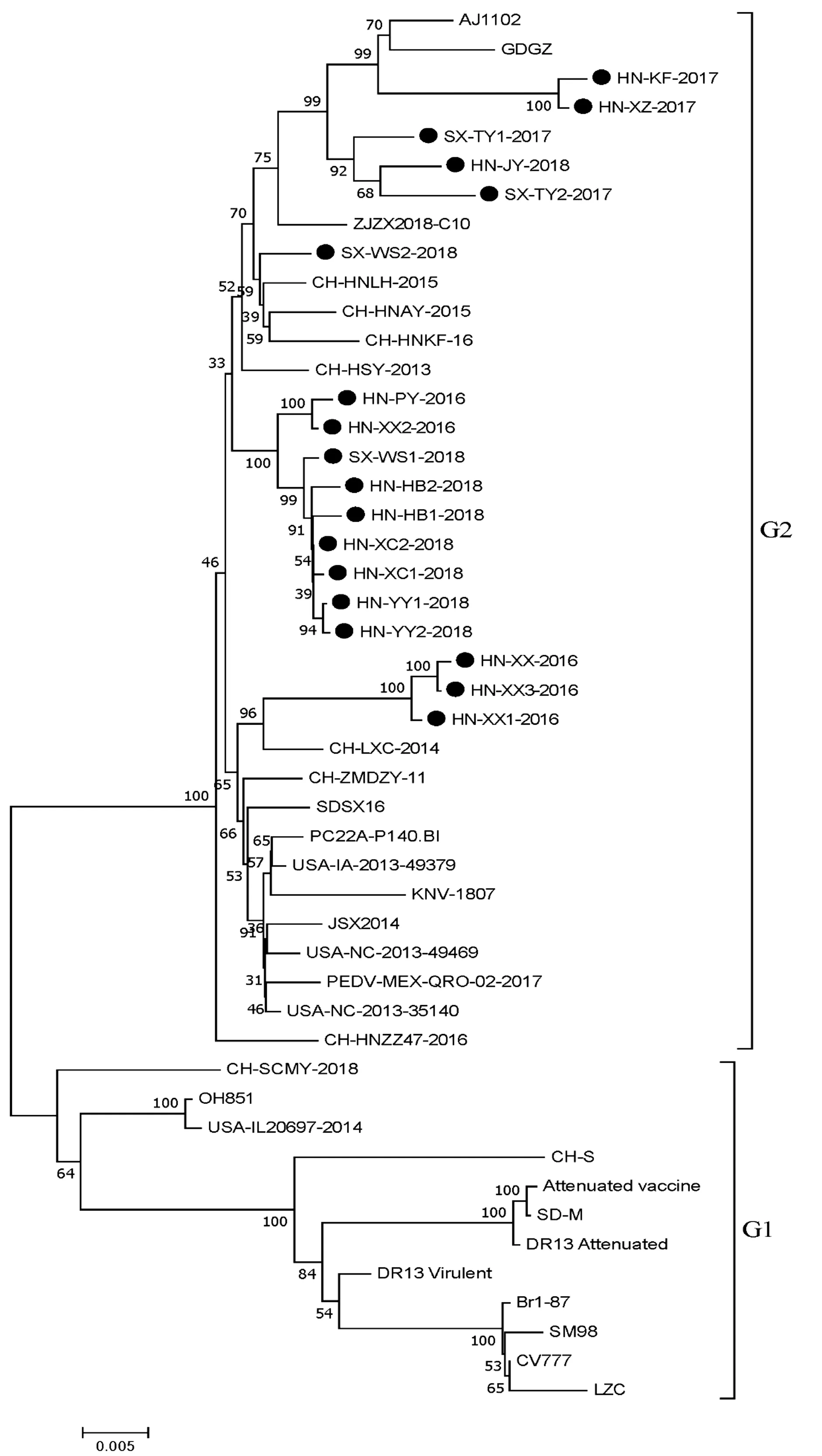
图3 PEDV S全基因遗传进化树 注:●表示本实验室分离毒株序列
通过对S基因编码氨基酸突变结果表明,与30株参考株相比,存在多处的点突变、碱基插入和缺失。这些点突变、碱基插入和缺失会改变氨基酸表达,是产生新型变异毒株的主要原因。同源性分析结果表明,18株PEDV S全基因间的同源性为96.0%~99.9%,与参考株S基因同源性为92.8%~99.3%。与河南往年分离株的同源性为96.2%~99.3%,与经典CV777毒株的核苷酸同源性为93.2%~93.7%,与美国新型毒株USA/IL20697/2014同源性为93.8%~96.0%。由此可见,近年来,从S基因的核苷酸和氨基酸的水平上看,PEDV S基因的突变主要集中在S1区域,本试验得到的18条流行株S全基因均在164~166 nt处有3个碱基(TTG)插入,177~185 nt有9个碱基(GGGTGTCAA)的插入,在427~429 nt有3个碱基(AAT)插入,在487~492 nt有6个碱基(CGTGAT)缺失。这几处的变异是与传统毒株区别,其毒力增强的主要原因。

图4 RDP4对PEDV S全基因重组分析
将本试验获得的18株S全基因核苷酸序列与GenBank中的30条PEDV参考株序列进行演化关系分析,结果将所有毒株分为2个群。G1群包括早些年分离得到的经典毒株序列以及疫苗株;G2群包括本试验扩增的18株毒株以及其他参考毒株,主要是2010年以后分离得到的流行株。与近几年的河南地区PEDV毒株均处在统一分支,具有较近的亲缘关系,这与赵丽等[16]和乔涵等[17]对河南地区PEDV S 基因序列分析的结果一致。本试验扩增的18株毒株与近几年的河南地区PEDV毒株以及国内外的流行株均处在同一分支,具有较近的亲缘关系。但是,在G2群内,又分出一些小分支,其中HN/KF/2017、HN/XZ/2017、SX/TY1/2017、HN/JY/2018和SX/TY2-2017与GDGZ、AJ1102处于同一小分支,核苷酸同源性为97.2%~99.6%,具有较近的亲缘关系。HN/XX/2016、HN/XX3/2016、HN/XX1/2016与CH/LXC/2014以及SX/WS2/2018与CH/HNLH/2015、CH/HNAY/2015、CH/HNKF/16和CH/HSY/2013分别处于一个小的分支,核苷酸同源性分别为98.2%~99.9%,98.5%~99.3%。本试验中的HN/PY/2016、HN/XX2/2016、SX/WS1/2018、HN/HB1/2018、HN/HB2/2018、HN/XC1/2018、HN/XC2/2018、HN/YY1/2018和HN/YY2/2018 9株毒株单独处于一个小分支,核苷酸同源性为98.9%~99.9%,亲缘关系较近,表明在2016—2018年间,PEDV S基因出现了一定的变异。
通过对S基因重组分析结果表明,克隆的SX/TY1/2017毒株S基因有一个重组开始位点位于3 291 nt,终止位点在4 132 nt位置,亲本株是CH/HNZZ47/2016和克隆的HN/KF/2017毒株。重组位点可能导致抗原表位发生变化,是病毒发生变异,产生新的族群的一种方式,也是免疫失败的原因之一[18-19]。值得注意的是,当前许多规模化猪养殖场所使用的PEDV疫苗免疫保护作用效果不佳,不能完全保护猪只免于感染发病,这与病毒抗原表位发生变异有很大关系。但是接种疫苗是实现疫病防控的关键环节,因此分析PEDV的遗传演化关系,正确认识现流行毒株与流行株、流行株与疫苗株之间的关系,筛选新的疫苗候选株,为开发新型疫苗防控PEDV具有重要意义。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
科学大观园(2022年2期)2022-01-23
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
动物医学进展(2015年10期)2015-12-07
特产研究(2014年4期)2014-04-10
