
POR基因突变致先天性肾上腺皮质增生症两家系遗传学分析
2020-09-17 08:50:38李莉刘玲毛会英
海南医学 2020年17期
李莉,刘玲,毛会英
1.泸州市妇幼保健院儿科,四川 泸州 646000;2.广东医科大学附属第三医院儿科,广东 佛山 528000
p450 氧化还原酶(p450 oxidoreductase,POR)基因位于7号常染色体长臂(Chr.7q11.2),长约7.1 kb,共16个外显子,编码680 个氨基酸组成黄素蛋白(NCBI Reference Sequence: NG_008930.1)[1],并参与电子传递反应。POR基因突变可影响21-羟化酶和17α-羟化酶的活性而导致p450 氧化还原酶缺陷症(p450 oxidoreductase deficiency,PORD)[2],其临床主要表现为骨骼发育不良(skeletal dysplasia)、肾上腺功能紊乱(adrenal dysfunction)、性发育异常(disorders of sex development,DSD)以及母孕期的女性男性化[3-5],先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是少见类型之一,为常染色体隐形遗传病[6],目前对PORD 病例报道较少。本研究对21-羟化酶A2、11-羟化酶B1 及17-羟化酶A1 基因无异常的CAH 患者进行POR 基因检测,为临床诊断提供依据,提供遗传咨询。
1 资料与方法
1.1 一般资料 选取2019年1~12月泸州市妇幼保健院及广东医科大学附属第三医院儿科门诊确诊的CAH患儿,收集21-羟化酶A2、11-羟化酶B1及17-羟化酶A1基因正常的2例患儿临床资料,并对其家系进行POR基因检测。
1.2 方法 经家长知情同意并签字后,留取患儿及其父母静脉血2 mL。提取全血基因组DNA 和RNA,设计14 对引物对POR 基因的16 个外显子及其侧翼进行PCR 扩增后Sanger 测序,上下游引物如表1所示。结果与GenBank中POR基因序列进行比对,并在其家属中进行验证。根据突变位置设计RNA引物,进行RNA逆转录后进行测序,测序结果在GenBank中进行比对,进一步验证突变位点,RNA 引物:For1,5'-CCACCTCATGCACCTGGAAT-3';Rev1,5'-CCCGGTACAGGTAGTCCTCA-3'。

表1 POR基因各外显子PCR扩增引物
2 结果
2.1 临床资料 先证者1,男性,1个月13 d,以体质量不增为首发症状,起病时间早,伴呕吐、腹泻,查体发现生殖器畸形(尿道下裂-会阴型),全身皮肤轻微色素沉着,双侧阴囊色素沉着明显(图1 A),辅助检查:电解质提示低钠高钾,17OHP 升高、醛固酮降低等(表2),染色体检查为正常男性核型。先证者2,男性,2岁11个月,以“阴茎短小”为主要表现(图1 B),无腹泻、呕吐、体质量不增等症状,查体发现阴茎外观短小,总长约4.5 cm,包埋部分约2.0 cm,阴茎弯曲,睾丸容积较同龄儿偏大(表2),辅助检查见表3。
2.2 基因检测结果
2.2.1 先证者1 外显子13,c.1508C>T (纯合突变),该突变导致POR 第503 位丙氨酸突变为缬氨酸(Valin,V/Val,GCC→GTC,p.A503V),如图2。该突变遗传自先证者1父母。
2.2.2 先证者2 外显子15,c.1820A>G (纯合突变),导致POR 第607 位氨基酸由酪氨酸(tyrosine,Y/Tyr)突变为半胱氨酸(cysteine,C/Cys,TAC→TGC,p.Y607C),如图3。该突变遗传自先证者2父母。
2.2.3 POR基因突变 POR基因外显子13,c.1508C>T 及外显子15,c.1820A>G 经Alamut Visual 及Mutatior Taster 功能软件预测会影响蛋白结构域的功能,通过Phyre2 功能软件可知503 位丙氨酸及607 位酪氨酸位于相对保守区域,如图4。

图1 两位先证者生殖器检查

表2 两位先证者的实验室检查
2.3 治疗 先证者1 给予糖皮质激素(氢化可的松片)、盐皮质激素(9α-氟氢化可的松)及氯化钠治疗,2 周后血钠、血钾恢复正常;先证者2给予糖皮质激素(氢化可的松片)治疗。

表3 两位先证者的临床评估
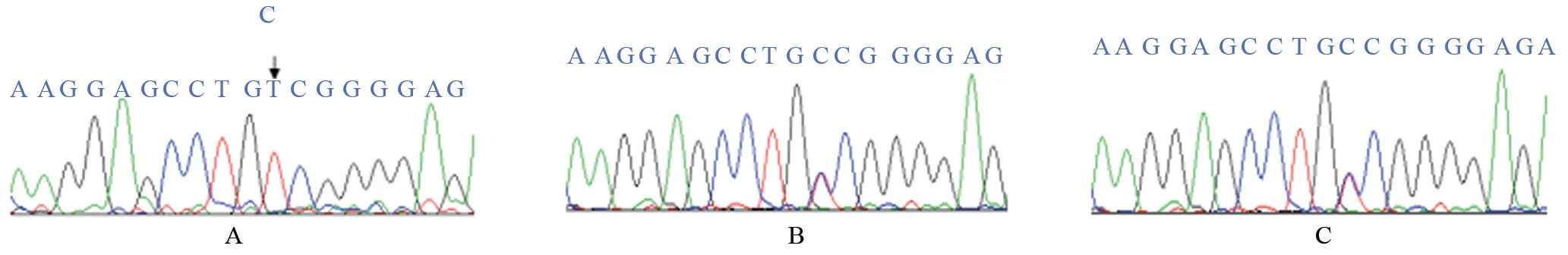
图2 外显子13基因测序图

图3 外显子15基因测序图

图4 POR突变图
3 讨论
CAH 是由于编码肾上腺皮质激素合成酶的一组基因发生突变,导致酶缺陷继而引起皮质激素合成不足,继发垂体促肾上腺皮质激素(adreno-cortico-tropic-hormone,ACTH)和下丘脑促肾上腺皮质激素释放激素(corticotropin releasing hormone,CRH)分泌增加,最终导致肾上腺皮质增生的常染色体隐形遗传病。CAH除了常见的21-羟化酶缺乏症(21-OHD)外,还有11α-羟化酶缺乏症(11α-OHD)、17-羟化酶缺乏症(17-OHD)、类脂性肾上腺皮质增生症(StAR)和p450氧化还原酶缺陷症(PORD)等少见类型。POR 是内质网中类固醇和药物代谢细胞色素p450 蛋白的代谢反应所必需的酶,是两种主要的类固醇生成酶17-羟化酶和21-羟化酶的电子供体[7]。POR突变使p450酶系活性受到影响,最终影响药物的代谢及类固醇激素的合成,导致一系列复杂的疾病,类似于多种类固醇代谢酶的联合缺陷[8-9]。
本研究中先证者1临床表现及实验室检查结果类似21-OHD 经典型,但21-羟化酶A2 基因无异常。POR基因提示外显子13,c.1820A>G(p.A503V,纯合突变),该突变来自先证者父母(杂合突变)。该位点为POR基因常见突变位点,影响类固醇合成酶及药物代谢酶活性。研究发现,c.1820A>G 突变使17-羟化酶与21-羟化酶活性分别降低32%与20%,并使17、20裂解酶活性降低42%[10-11],导致皮质醇和醛固酮合成受损,皮质醇低下,经负反馈使ACTH分泌增加,刺激肾上腺皮质细胞增生,以增加皮质醇合成,但17-羟化酶与21-羟化酶活性降低使皮质醇依然低下,且孕酮转化为醛固酮降低,导致水盐平衡失衡;但雄激素合成通路正常,在高ACTH刺激下,堆积的17-OHP和孕酮向雄激素转化增多,产生了旁路代谢亢进-高雄激素血症(雄烯二酮、睾酮和脱氢表雄酮升高),临床表现与21-OHD 相似。先证者2,外显子15,c.1820A>G (p.Y607C,纯合突变),该突变来自先证者父母(杂合突变)。POR 第607 位酪氨酸位于烟酰胺腺嘌呤二核苷酸磷酸(NADPH)结合域,酪氨酸的羟基和NADPH 第一正磷酸基团之间形成氢键,使NADPH 和黄素腺嘌呤二核苷酸(FAD)紧密结合,传递电子[12-13]。POR 第607 位酪氨酸突变为半胱氨酸改变了NADPH 与FAD的结合,从而使POR活性下降60%~90%[14-15],抑制17-羟化酶、21-羟化酶与19-羟化酶活性,但以19-羟化酶为主,皮质醇和醛固酮通路基本正常,睾酮转化为雌二醇通路受阻,使其不能转化为雌二醇,导致高雄激素血症,因此先证者2 临床表现较先证者1 轻。本研究两位先证者均为纯合突变,突变均来源其杂合突变的父母,符合常染色体隐性遗传规律。
PORD 的临床表现联合了21-OHD 和17-OHD,根据不同突变对酶活性的影响程度,PORD的临床表现多样,容易导致临床上CAH患者由于临床表现、体征和实验室检查不典型而出现误诊漏诊。PORD 治疗同21-OHD,最重要的是纠正肾上腺皮质功能减退危象,维持机体正常的生理代谢,降低死亡率并且抑制垂体ACTH 分泌,延缓肾上腺雄激素过度分泌导致的性早熟。治疗不当与治疗过度均可导致矮小及生理、心理发育障碍等后遗症。因此定期随访很重要,根据临床症状及检查结果及时调整治疗方案,以最低药物剂量达到最好的代谢控制,避免或减少药物副作用,使患者能达到正常的生长及青春发育,提高生活质量。
本研究通过对2例PORD患儿的临床表现和基因突变特点分析,POR 基因外显子13,c.1508C>T、外显子15,c.1820A>G突变是这两例患者的致病原因,提高了临床医师对本病的认识,并扩大了基因突变谱,为遗传咨询提供了基础。
猜你喜欢
中华实用诊断与治疗杂志(2022年2期)2022-09-02 01:47:02
临床输血与检验(2022年3期)2022-06-22 02:52:50
热带作物学报(2020年9期)2020-10-29 07:35:39
中国现代医生(2020年12期)2020-07-04 03:03:59
家庭医学(下半月)(2019年9期)2019-10-12 08:03:58
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
天然产物研究与开发(2018年1期)2018-02-02 07:21:22
中成药(2018年1期)2018-02-02 07:19:57
中国塑料(2016年7期)2016-04-16 05:25:52
