
铁铈氧化物催化环己酮氧气氧化制ε-己内酯
2020-09-15 08:00王媛媛胡珊珊刘赛赛朱明乔
高校化学工程学报 2020年4期
王媛媛, 张 明, 胡珊珊, 刘赛赛, 朱明乔
(1. 浙江大学 化学工程与生物工程学院, 浙江 杭州 310027; 2. 台州市源众药业有限公司, 浙江 台州 318000)
1 前 言
ε-己内酯(ε-caprolactone, ε-CL)是一种用途广泛的有机合成中间体,常用于合成聚己内酯(polycaprolactone, PCL),PCL 具有较好生物相容性、可降解性、热塑性等,可制成环保材料与生物医用材料[1-3]。ε-己内酯主要通过环己酮(Baeyer-Villiger, B-V)氧化重排反应得到,工业常用有机过氧酸作氧化剂,如过氧乙酸有氧化能力强、来源丰富等优点,但也存在储存不安全、腐蚀设备和产生废酸等不足[4]。采用氧气作为氧化剂代替传统过氧酸,具有绿色清洁、廉价安全等优势[5],但分子氧氧化能力较弱,常需加入催化剂以及醛类助氧化剂来提高反应活性,目前效果最优的醛为苯甲醛,高效非均相催化剂的研究也成为热点。近年来报道的催化剂主要有以Cu、Co、Mn 等过渡金属元素为活性组分的分子筛型催化剂(如Cu-MCM-41[6-7]),金属氧化物型催化剂(如SnO2、TiO2、CuO 等一种或多种的混合物[8-9])以及石墨烯、纳米二氧化硅等非金属氧化物型催化剂[10-11],但这些催化剂常存在制备周期长、活性组分流失快、活性不高、价格昂贵等问题。本研究选择铁铈双金属氧化物作催化剂,具有制备简单、高效且易回收等优点,Fe2O3能够活化苯甲醛的醛基和环己酮的羰基;而CeO2对活性氧有优越的储存和传递作用,近年来在CO 氧化、光催化氧化、电催化氧化、有机物裂解等研究中均有应用[12-15],但在环己酮氧化上鲜有应用。采用共沉淀法制备一系列不同铁铈摩尔比及不同焙烧温度的催化剂,对催化剂的结构、形貌等进行表征,并考察其在分子氧氧化环己酮制备ε-己内酯反应中的催化效果。
2 实验(材料与方法)
2.1 实验原料与催化剂制备
六水合硝酸亚铈(Ce(NO3)3·6H2O, 99.95%)购于上海阿拉丁生化科技股份有限公司;九水合硝酸铁(Fe(NO3)3·9H2O;≥98.5%)、氨水(NH3·H2O,25%~28%)、碳酸氢铵(NH4HCO3,21%~22%)购于国药集团化学试剂有限公司,氧气、氮气购于杭州今工特种气体有限公司。不同铁铈摩尔比的氧化物(n(Fe):n(Ce)= 1:4~3:1)通过共沉淀法合成,称取一定量Ce(NO3)3·6H2O 和Fe(NO3)3·9H2O,溶于50 mL 去离子水中,40 ℃搅拌溶解。称取等摩尔量的NH3·H2O 和NH4HCO3,用蒸馏水溶解并稀释成NH4+浓度为1 mol·L-1的混合液,在剧烈搅拌下缓慢滴入盐溶液中,直至溶液pH 为9.5,保温搅拌2 h 后将反应液降至室温老化48 h。沉淀通过抽滤洗涤至中性,于80 ℃干燥12 h,将前驱体置于管式炉中以5 ℃·min-1升温至600 ℃,焙烧3 h 得一系列FeCe (x:y-600)氧化物,选定铁铈比为1:2,改变焙烧温度为500、400、300 ℃得催化剂FeCe (1:2-T)(x:y 表示原料铁盐和铈盐的摩尔比,T 指焙烧温度),采用单一金属盐进行沉淀,500 ℃焙烧可得CeO2和Fe2O3。
2.2 催化剂表征
催化剂晶相结构分析采用荷兰帕纳科公司的X’Pert PRO 型X 射线衍射仪,CuKα 靶作为辐射源,X射线波长λ=1.540 6 nm,阶宽为0.02°,扫描范围为2θ=10°~80°;催化剂形貌分析采用日本日立公司的SU-8010 型扫描电子显微镜,加速电压0.1~30 kV,观测倍率20~80 000 倍;催化剂比表面积和孔结构测试采用美国Micromerities Tristar ASAP 2020 型全自动比表面及微孔、介孔物理吸附分析仪,样品于200 ℃脱附3 h 后,在-196 ℃用N2物理吸附法进行测试;样品表面组成分析采用Thermo Scientific 公司ESCALAB 250Xi 型X 射线光电子能谱仪,AlKα 靶作为光源。
2.3 催化剂性能评价
环己酮氧化反应在带冷凝管的三颈烧瓶中进行,依次加入100 mg 催化剂,12 mmol 环己酮,24 mmol苯甲醛及20 mL 1,2-二氯乙烷,搅拌升温至50 ℃,接着用质量流量计以10 mL·min-1鼓入O2,恒温反应3 h,离心分离出催化剂,溶剂反复洗涤后烘干回收。反应液以正庚烷作内标,通过GC 1690 气相色谱仪检测,SE-54 毛细管柱,进样口和检测器温度为270 ℃,柱箱初温60 ℃保留3 min,以10℃·min-1升温至250 ℃保留3 min。
3 实验结果与讨论
3.1 催化剂表征分析
3.1.1 催化剂的XRD 分析
不同铁铈摩尔比的样品X射线衍射图如图1所示,样品FeCe(3:1-600)~FeCe(1:4-600)在2θ = 28.5°(111)、33.1°(200)、47.5°(220)、56.3°(311)处的衍射峰归属于具有立方萤石结构的CeO2(PDF# 34-0394),在2θ = 24.2°(012)、33.2°(104)、35.6°(110)、40.9°(113)处的衍射峰归属于α-Fe2O3(PDF# 33-0664),且随着铁含量的减少,α-Fe2O3的衍射峰强度逐渐减弱,CeO2的衍射峰强度逐渐增强,证明不同铁铈比例的氧化物合成成功。铁铈氧化物中α-Fe2O3的特征峰变得很弱,可能是由于Fe2+(0.075 nm) 和 Fe3+(0.064 nm) 离子半径小于Ce4+(0.097 nm),使部分Fe2O3能够进入CeO2的晶格结构形成固溶体,这不仅让催化剂的结构变得更加稳定也更容易形成氧缺陷位[16]。

图1 不同铁铈摩尔比下FeCe(x:y-600)的XRD 图Fig.1 XRD patterns of FeCe (x:y-600) under different Fe/Ce molar ratios
3.1.2 催化剂的BET 分析
为了探究焙烧温度对催化剂结构的影响,取FeCe(1:2-400)、FeCe(1:2-500)、FeCe(1:2-600)进行N2吸附-脱附测试。由表1 数据可知,焙烧温度为400 与500 ℃时催化剂结构参数相差不大,600 ℃时比表面积和孔容大大下降,这是由于高温焙烧使催化剂的结晶度得到很大提高,催化剂表面趋于光滑、内部出现烧结导致孔道封闭。由图2(a)可以看出,3 种催化剂的吸附脱附等温线属于第IV 类等温线,在p/p0为0.55~1.0 时均伴有H3型滞后环,表明材料具有片状物松散堆积而形成的楔形孔。结合图2(b)可知,FeCe(1:2-400)在孔径为2~3 nm 处分布着少量较小的孔,在3~15 nm 处分布着大量楔形孔;FeCe(1:2-500)同样连续分布着孔径为2~4 nm 及4~18 nm 两类孔;FeCe(1:2-600)的孔径分布很宽,在2~4 nm 及8~50 nm 均存在孔,50 nm 以后可能存在大孔,大部分孔可能为光滑片层之间的空隙,而不是孔道。FeCe(1:2-400)、FeCe(1:2-500)两种催化剂孔径分布较窄,孔道尺寸均一,且比表面积和孔容较大,更有利于底物和催化剂充分接触。
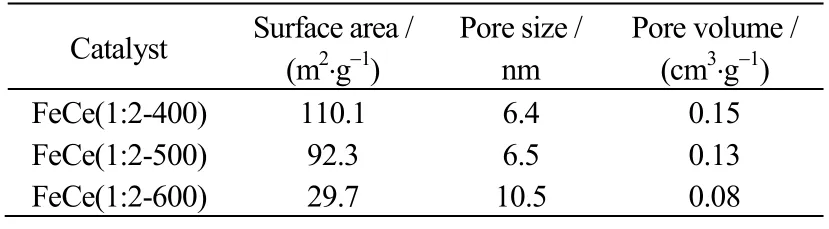
表1 催化剂的结构参数 Table 1 Structure parameters of the catalysts

图2 催化剂FeCe(1:2-T)的N2 吸附-脱附等温曲线和孔径分布 Fig.2 N2 adsorption desorption isotherms and pore size distribution of the FeCe(1:2-T) catalysts
3.1.3 催化剂的XPS 分析
3 个不同焙烧温度样品FeCe(1:2-T)和单一CeO2、Fe2O3的XPS 表征如图3 所示,分别对Fe、Ce、O元素的能谱图进行分峰计算得到催化剂表面元素相对含量,结果列于表2。图3(a)为Fe 元素的XPS 能谱图,以FeCe(1:2-400)为例,在710.70、716.60 eV 处分别为Fe2+的Fe 2p3/2的主峰和卫星峰,在712.60、719.80 eV处分别为Fe3+的Fe 2p3/2的主峰和卫星峰[17];Ce元素的XPS谱图如图3(b)所示,在y(882.40 eV)、y’’(888.80 eV)、y’’’(898.40 eV) 和w(900.90 eV)、w’’ (907.40 eV)、w’’’ (916.80 eV)处分别归属于Ce4+的Ce 3d5/2和Ce 3d3/2的特征峰,y’(884.60 eV)和w’ (902.66 eV) 归属于Ce3+的特征峰,说明样品表面存在Ce3+和Ce4+两种价态的铈[18];O 元素的XPS 能谱图如图3(c)所示,图中氧的峰较宽且带有肩峰,在529.55、531.45 eV 的峰分别代表金属氧化物的晶格氧(OLattice,如O2-)及表面吸附氧(OSurface,如O2-,O-),其他样品的谱图分峰位置跟FeCe(1:2-400)相近。从表2 元素价态比值可知单一CeO2表面Ce3+和吸附氧相对含量最低,Fe2O3表面Fe2+相对含量最高,结合XRD 分析结果可知FeCe(1:2-T)中Fe2O3能够进入到CeO2的晶格,推测Fe2+能掺入CeO2中,容易发生Ce4++Fe2+↔Ce3++ Fe3+的反应,同时可能会转化晶格氧成为表面氧(Ce4+↔ Ce3++[O]),在结构上形成氧缺陷位[19]。样品表面Ce3+相对含量随着焙烧温度升高有略微下降趋势,表面氧相对含量在500 ℃时达到最高,可能是因为适当的升温有利于掺杂过程进行,温度达600 ℃时颗粒变大,Fe2+的掺入量减少,表面Ce3+浓度降低。FeCe(1:2-500)较其他样品具有更高的表面氧浓度,同时具有适中的Fe2+、Ce3+表面浓度。
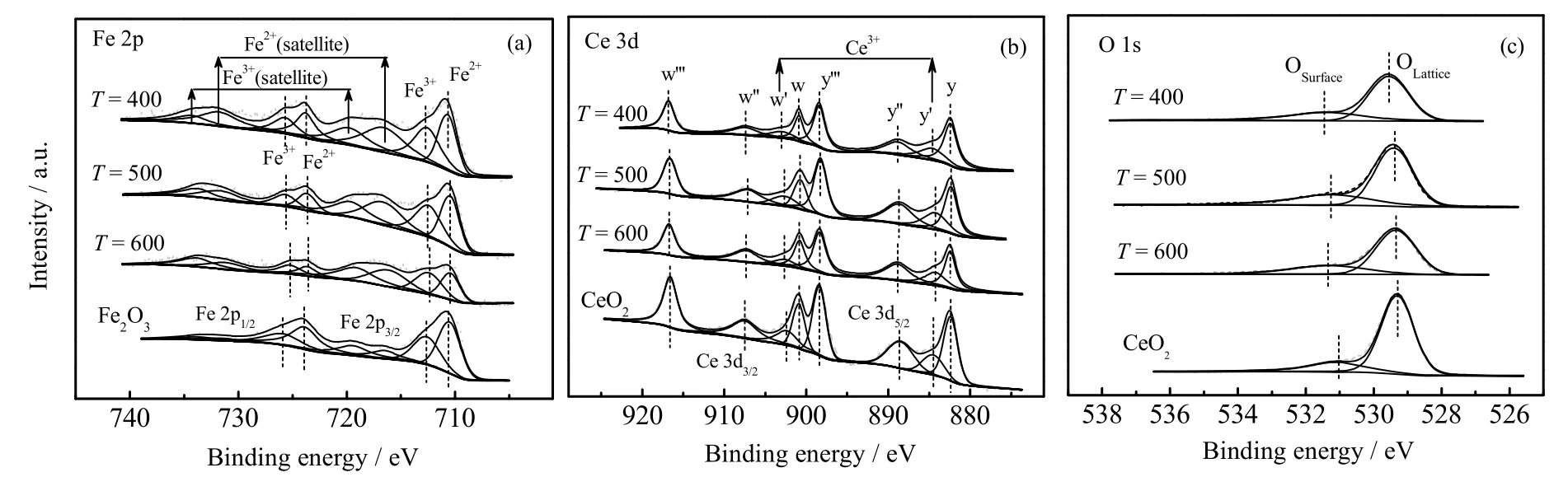
图3 催化剂FeCe(1:2-T)的XPS 谱图 Fig.3 XPS spectra of the FeCe(1:2-T) catalysts

表2 催化剂XPS 数据总结 Table 2 Summary of XPS data of the catalysts
3.1.4 催化剂的SEM 分析
图4 为不同焙烧温度下FeCe(1:2-T)的扫描电镜图,焙烧温度对催化剂形貌影响较大:(a) FeCe(1:2-600)由不规则片层结构堆叠而成,片层表面较为平滑;(b) FeCe(1:2-500)中片层结构仍然清晰,部分片层成为小碎片;(c) FeCe(1:2-400)中为块层且更为细碎,表面颗粒感更为明显;(d) FeCe(1:2-300)由细小的球状颗粒团聚形成的不规则球形和块状结构组成。这表明催化剂的结构随焙烧温度降低发生较为规律性的变化,催化剂的比表面积随之增大,结晶度随之降低,这也与BET 分析结果相符。较高的结晶度能令催化剂具有高结构稳定性,而较大比表面积对催化环己酮氧化更为有利,因此合适焙烧温度的选择十分重要。

图4 不同焙烧温度下FeCe(1:2-T)的SEM 图 Fig.4 SEM images of FeCe(1:2-T) under different calcination temperatures
3.2 催化剂在环己酮氧气氧化反应中的性能
环己酮分子氧氧化制备ε-己内酯的过程符合自由基机理:苯甲醛在催化剂和O2作用下经自由基转移形成过氧苯甲酸,接着与环己酮发生亲核加成,发生重排并脱去苯甲酸得到产物ε-己内酯。一系列铁铈催化剂在环己酮B-V 反应中的催化效果如表3 所示,反应条件一定时,催化剂的加入能够显著提高反应活性。其中,FeCe(x:y-T)催化活性优于单一的Fe2O3或CeO2,可能是双金属氧化物催化时存在协同作用,使环己酮和苯甲醛的羰基更容易被活化。铁铈摩尔比的改变会影响催化剂活性,铁含量的增加一定程度上有助于环己酮转化,当铁铈比高于1:1 后,ε-己内酯选择性出现下降,n(Fe):n(Ce)=1:2 时铁铈的协同作用最佳。焙烧温度的改变同样对催化活性造成影响,焙烧温度为500 ℃的催化剂FeCe(1:2-500)催化效果最佳,环己酮转化率为98.8%,ε-己内酯选择性大于99% ( entry 8)。综上所述,选择FeCe(1:2-500)进行环己酮氧化反应条件影响的考察。

表3 不同催化剂在环己酮B-V 氧化反应中的催化性能 Table 3 Catalytic performance of different catalysts in the B-V oxidation of cyclohexanonea
3.3 反应条件对FeCe(1:2-500)催化环己酮氧气氧化反应的影响
3.3.1 反应溶剂的影响
溶剂对于反应的影响如表4 所示,在一定条件下,溶剂对于环己酮的转化率影响很大。以非极性溶剂1,2-二氯乙烷为溶剂时反应效果最佳,环己酮的转化率达98.8%;以极性溶剂乙腈、乙酸乙酯为溶剂时,环己酮的转化率有所下降,但仍在80%以上,以甲苯和1,4-二氧六环为溶剂时反应活性大大降低,环己酮转化率低于10%。因此,选用1,2-二氯乙烷作为环己酮氧气氧化制备ε-己内酯的反应最为合适。

表4 溶剂对环己酮B-V 氧化反应的影响a Table 4 Effects of solvents on B-V oxidation of cyclohexanonea
3.3.2 催化剂用量的影响
为考察催化剂用量对环己酮B-V氧化反应的影响,仅改变催化剂用量,保持其余反应条件为环己酮12 mmol,苯甲醛24 mmol,1,2-二氯乙烷20 mL,O210 mL·min-1,50 ℃反应3 h 进行实验。结果如图5 所示,催化剂用量少于50 mg 时,环己酮的转化率随催化剂用量增加而显著增加,多于50 mg 后趋于平稳,反应选择性均大于99 %。这可能是由于催化剂活性位点与底物结合而降低反应活化能,随催化剂量增加,被活化的底物量也增加,反应物迅速转化;当催化剂量提供的活性位点过多时,反应转化率不再提高。催化剂用量为50 mg,环己酮转化率为97.7%,ε-己内酯选择性为99.5%,选择用量50 mg 进行下列的考察。

图5 催化剂用量对环己酮B-V 氧化反应的影响 Fig.5 Effects of catalysts dosage on B-V oxidation of cyclohexanone
3.3.3 反应时间的影响
保持其余反应条件不变,改变反应时间来考察其影响。反应结果如图6,环己酮转化率随反应时间逐渐升高,ε-己内酯选择性几乎不变,反应时间达3 h 后环己酮的转化率升幅较小。可能是因为反应初期原料浓度大使反应速率较快,且反应为自由基反应,在过量苯甲醛环境下,苯甲醛能够优先氧化形成自由基中间体,紧接着引发环己酮氧化,使反应的环己酮均转化为ε-己内酯,反应选择性很高。当反应4 h时,环己酮转化率达到99.2%,ε-己内酯的选择性为99.6%,其后反应趋于平衡,因此4 h 是比较合适的反应时间,选择4 h 作为反应时间进行下面的考察。

图6 反应时间对环己酮B-V 氧化反应的影响 Fig.6 Effects of reaction time on B-V oxidation of cyclohexanone
3.3.4 反应温度的影响
图7 为不同反应温度下环己酮B-V 氧化反应的结果图,在其他反应条件一定时,温度对于该反应无显著影响。反应为35 ℃时,环己酮转化率已达98.0%,说明FeCe(1:2-500)具有较高的催化活性,即使在较低温度下也能使反应物快速转化。转化率随温度升高有一定增加,幅度并不明显,直到45 ℃时已基本不变,当反应温度高达60 ℃时,反应选择性出现下降,可能是由于温度较高时ε-己内酯过度氧化成己二酸。因此,较为合适的反应温度为45 ℃,此时反应的转化率达99.2%,选择性为99.7%。
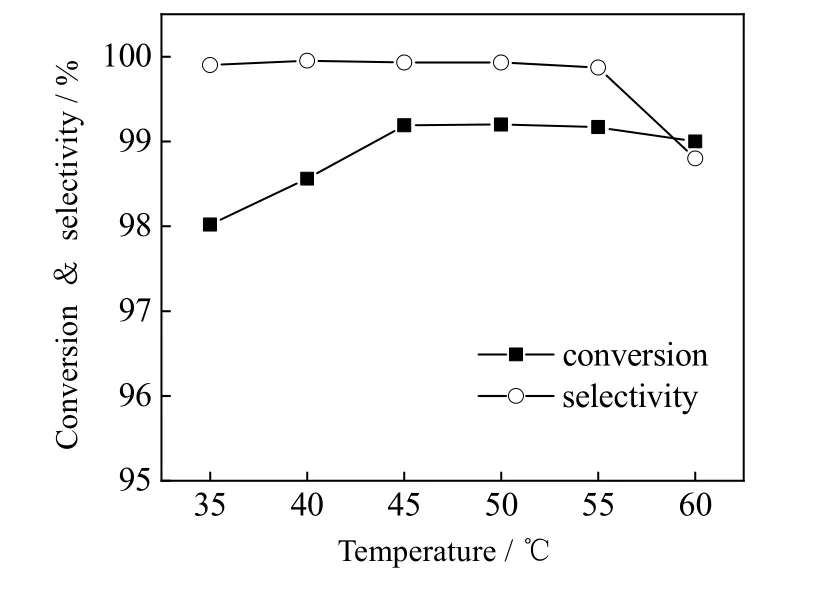
图7 反应温度对环己酮B-V 氧化反应的影响 Fig.7 Effects of reaction temperature on B-V oxidation of cyclohexanone
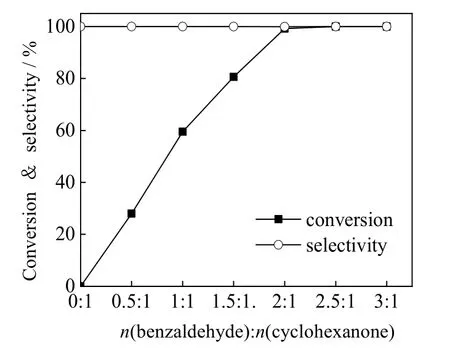
图8 苯甲醛与环己酮摩尔比对B-V 氧化反应的影响 Fig.8 Effects of molar ratios of benzaldehyde to cyclohexanone on B-V oxidation of cyclohexanone
3.3.5 苯甲醛和环己酮摩尔比的影响
苯甲醛作为反应的助氧化剂对环己酮B-V 氧化十分重要,通过改变苯甲醛与环己酮的摩尔比,探究醛酮比对反应的影响。由图8 可知,苯甲醛的用量会对反应转化率产生很大影响,醛酮摩尔比从0.5:1增加至2:1 的过程中,环己酮转化率显著提高,苯甲醛用量继续增加,转化率增加已不明显。理论上应只需与环己酮当量的苯甲醛就能令环己酮完全转化,但实际上醛酮摩尔比为2:1 才可达到,可能是因为苯甲醛在O2和催化剂作用下产生含氧自由基中间体速度很快且很不稳定,而环己酮羰基活化较慢,不能很快接收中间体传递的氧,因此需要更多的苯甲醛才能实现环己酮的完全转化。从反应结果得出,苯甲醛与环己酮的摩尔比仍为2:1 较为合适。
3.4 催化剂的重复性
实验证明催化剂FeCe(1:2-500)在环己酮O2氧化制备ε-己内酯的反应中表现出良好催化性能,但催化剂需得到进一步应用就要具备稳定的重复催化效果。首次反应后催化剂通过离心分离、溶剂反复洗涤及100 ℃干燥后回收,重复进行氧化反应。从表5 可知,催化剂重复使用5 次,效果无明显下降,表示该催化剂具有较好的结构稳定性,有一定的工业应用前景。

表5 回收催化剂的催化性能a Table 5 Catalytic performance of the recycled catalyst a
4 结 论
采用共沉淀法合成不同铁铈摩尔比和焙烧温度的催化剂FeCe(x:y-T),并用于催化O2/苯甲醛共氧化的环己酮B-V 氧化反应。BET 和SEM 表征结果说明,催化剂由片层结构堆积形成,焙烧温度为500 ℃时晶形完整且比表面积较大。XRD 和XPS 表征说明,部分Fe2O3会进入CeO2晶格,让CeO2形成氧缺陷位,焙烧温度为500 ℃时催化剂具有最多的表面吸附氧。环己酮B-V 氧化反应结果表明,改变铁铈摩尔比会让催化剂活性产生规律性变化,双金属氧化物的协同效应使催化效果优于单一金属氧化物,筛选出最优催化剂为FeCe(1:2-500)。反应条件优化后,得到合适的条件为n(环己酮):n(苯甲醛)=1:2,环己酮12 mmol,催化剂50 mg,1,2-二氯乙烷20 mL,45 ℃反应4 h,反应转化率和选择性均大于99%,且催化剂稳定性较高,重复使用5 次无明显活性下降。
猜你喜欢
化学反应工程与工艺(2022年1期)2022-04-23
化工管理(2021年7期)2021-05-13
陶瓷学报(2020年6期)2021-01-26
天津中医药(2020年5期)2020-06-01
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
化学反应工程与工艺(2017年6期)2017-06-12
中成药(2017年4期)2017-05-17
中国民族医药杂志(2016年6期)2016-05-09
郑州大学学报(工学版)(2015年6期)2015-03-18
