
利用2个F2群体整合中国豌豆高密度SSR遗传连锁图谱
2020-09-14 07:22张红岩黄宇宁季一山徐东旭郭瑞军宗绪晓
作物学报 2020年10期
刘 荣 王 芳 方 俐 杨 涛 张红岩 黄宇宁 王 栋,3 季一山 徐东旭 李 冠 郭瑞军 宗绪晓,*
利用2个F2群体整合中国豌豆高密度SSR遗传连锁图谱
刘 荣1,**王 芳1,**方 俐1,**杨 涛1张红岩1黄宇宁1王 栋1,3季一山1徐东旭2李 冠1郭瑞军1宗绪晓1,*
1中国农业科学院作物科学研究所作物种质资源中心, 北京 100081;2张家口市农业科学院食用豆类研究所, 河北张家口 075000;3山东省作物种质资源中心, 山东济南 250100
豌豆(L.)是一种重要的食用豆类作物, 在全世界范围内广泛种植, 既可作为人类食物, 也可作为牲畜饲料。用SSR标记构建的遗传连锁图谱在豌豆和其他作物的标记辅助育种中发挥着重要的作用。尽管对豌豆遗传连锁作图的研究已有悠久历史, 但公众可获得且可转移的SSR标记以及基于遗传独特的中国豌豆种质的高密度遗传连锁图谱仍然有限。为了获得更多可转移的SSR标记和中国豌豆的高密度遗传连锁图谱, 本研究首先从自主开发和文献获取的12,491个全基因组SSR标记中筛选了617个多态性SSR标记, 并用于G0003973×G0005527 F2群体遗传连锁图谱的加密。加密后的图谱全长扩展到5330.6 cM, 包含603个SSR标记, 标记平均间距离8.8 cM, 相比之前的图谱有明显改善。基于上述结果, 我们又筛选了119个具有多态性的SSR标记, 用于构建大样本W6-22600×W6-15174 F2群体的遗传连锁图谱, 新图谱累积长度为1127.1 cM, 包含118个SSR标记, 装配在7条连锁群上。最后, 将来自以上2个遗传图谱的数据进行整合, 得到了一张覆盖范围6592.6 cM的整合图谱, 包含668个SSR标记, 由509个基因组SSR、134个EST-SSR和25个锚定标记组成, 分布在7条连锁群上。这些SSR标记和遗传连锁图谱将为豌豆的遗传研究和标记辅助育种提供有力工具。
豌豆; SSR; 遗传连锁图谱; 整合图谱; 标记辅助育种
豌豆(L.)属于豆科(Leguminosae/ Fabaceae), 野豌豆族(Vicieae), 豌豆属(), 染色体数为2= 2= 14, 基因组大小约为4.45 Gb[1-2]。豌豆富含蛋白质和多种营养元素, 是经济上最重要的食用豆类作物之一, 在世界范围内广泛种植, 既可以作为谷物和蔬菜供人类食用, 又可作为牲畜的饲料[3-4]。根据FAO的统计, 2018年, 豌豆(包括青豌豆和干豌豆)在全球食用豆类作物中的总产量仅次于普通菜豆; 同时, 中国青豌豆的总产量居世界首位, 而干豌豆的总产量仅次于加拿大和俄罗斯[5]。此外, 豌豆因其固氮能力而被认为是一种环境友好型作物, 在可持续农业系统中起着至关重要的作用[6]。
高密度遗传连锁图谱是功能基因定位、比较基因组学以及分子辅助育种等研究的重要工具[7]。以前, 人们一直致力于利用包括RFLP、RAPD、SSR和SNP在内的多种分子标记基于不同类型的群体来构建豌豆的遗传连锁图谱[8-15]。最近, 有学者针对豌豆开发了基于高密度SNP的遗传连锁图谱, 并为鉴定重要农艺性状的遗传基础提供了强大的工具[16-19]。此外, 新近公布的豌豆参考基因组也为理解豌豆关键农艺性状的分子基础并促进其育种改良奠定了重要基础[20]。
SSR标记因其具有信息量丰富、共显性遗传、多等位基因、基因组覆盖广等特性, 同时在相近物种之间具有可重复性和可移植性[21-22], 在遗传多样性评估和物种亲缘关系鉴定[23-24]、遗传连锁图谱构建[25-26]、标记辅助选择[27-28]、DNA指纹图谱鉴定[29-30]等方面具有显著优势。相比基因组SSR, 位于基因区的EST-SSR因其具有更高的可转移性、较低的开发成本以及与基因的密切关系, 而越来越受到人们的重视[11,21]。然而, 尽管针对豌豆的遗传连锁作图研究已有很长的历史, 并且在豌豆中已经构建了几十种具有不同标记的遗传连锁图谱[31], 但公众可获得的可用于豌豆遗传研究的SSR上图标记较少, 同时基于遗传独特的中国豌豆种质[1,32-33]的遗传连锁图谱仍然有限。值得注意的是, 过去基于中国豌豆种质构建的遗传连锁图谱包括157个SSR标记, 分布在11个连锁群中, 全长1518 cM, 标记数量较少, 需要进一步加密并完善至7个连锁群[15]。
与单个遗传连锁图谱相比, 整合遗传连锁图谱由于整合了多个群体的信息而具有多种优势[34-35], 例如具有更高的标记密度, 更完整的基因组覆盖范围, 可对不同群体进行标记共线性比较等, 在许多作物包括豌豆中均有应用[16,20,36-37]。因此, 本研究的目的如下: 1)筛选豌豆中可移植转换的SSR标记, 用于豌豆的遗传研究和分子作图。2)对我们以往基于G0003973×G0005527 F2群体, 构建的遗传连锁图谱进行加密。3)基于W6-22600×W6-15174 F2群体, 构建新的遗传连锁图谱。4)结合上述2个基于中国种质的遗传连锁图谱信息, 构建一张豌豆整合SSR遗传连锁图谱。
1 材料与方法
1.1 作图群体
本研究利用基于中国豌豆种质为亲本的2个F2群体进行遗传连锁作图。群体1 (PSP1)与本实验室之前的研究相同[15], 来自母本G0003973 (耐寒)和父本G0005527 (不耐寒)之间的杂交, 由190个F2个体组成。群体2 (PSP2)则是以母本W6-22600 (多小叶)与父本W6-15174 (无小叶)进行杂交, 由480个F2个体组成。
1.2 SSR标记筛选
利用本实验室自主开发[38]和文献获取[39-41]的12,491个SSR标记(包括11,145个基因组SSR和1346个EST-SSR), 对PSP1的亲本及随机选择的4个F2个体进行全基因组扫描, 筛选出多态性SSR标记用于遗传连锁作图。此外, 从以往研究中已发表的豌豆遗传连锁图谱中, 选择具有已知连锁群位置的125个SSR标记[10,42-44], 利用PSP1和PSP2的亲本和4个随机选择的F2个体来筛选锚定标记。
1.3 DNA提取和PCR扩增
在2个F2群体种植当年, 收集每个F2个体植株的嫩叶, 经液氮速冻后, 使用改良的CTAB方法[45]提取基因组DNA。用NanoDrop 2000检测DNA浓度并稀释到工作液浓度50 ng μL-1后, 于-20℃保存备用。
PCR扩增反应体系为10 μL, 包含1.5 μL基因组DNA (50 ng μL-1)、5 μL 2×PCR Master Mix (Genstar, 中国北京)、0.5 μL正向引物 (2 μmol L-1)、0.5 μL反向引物 (2 μmol L-1)和2.5 μL ddH2O。PCR产物通过8%非变性聚丙烯酰胺凝胶电泳(PAGE)分离, 并通过0.1%硝酸银染色。根据片段大小记录等位基因状态, SSR标记状态编码如下: 与父本相同的带型记为“AA”, 与母本相同的带型记为“BB”; 具有双亲带型的记为“AB”; 缺失或无效的带型记为“-”。只有那些能够扩增出清晰条带并可以显示亲本多态性的SSR标记才被选择用于后续的基因分型。
1.4 遗传连锁图谱构建
分别对PSP1和PSP2的所有F2个体进行基因分型, 并去除缺失数据超过20%的标记或个体。使用χ²分析来检测标记偏分离状况, 并使用Bonferroni校正对= 0.05的显著性水平进行校正, 在进一步的遗传作图中排除显著偏分离的标记。利用Kosambi作图函数对2个群体构建遗传连锁图谱, LOD>2。在以往公布的豌豆遗传连锁图谱的基础上, 通过筛选得到的锚定标记对每个连锁群进行分组[10,42-44]。然后, 利用共有标记将这2个群体的信息整合到一张遗传连锁图谱上。以上所有分析均利用QTL IciMapping V4.0软件完成[46]。遗传连锁图谱和物理图谱利用MapChart V2.3软件进行可视化展示[47]。然后, 本研究以新近发表的豌豆基因组为参考(Caméor genome build 1a)[20], 利用KnowPulse网站(https:// knowpulse.usask.ca/blast/nucleotide/nucleotide)的BL ASTn工具对50个共有标记的扩增片段序列进行比对, 参数选取默认参数, E-value设为1e−3。
2 结果与分析
2.1 多态性标记筛选
利用本实验室自主开发[38]和文献获取[39-41]的12,491个SSR标记(包括11,145个基因组SSR和1346个EST-SSR), 对PSP1的亲本及随机选择的4个F2个体进行全基因组扫描, 初步筛选出扩增条带清晰且在父母本间呈多态性差异的954个多态性SSR标记, 用于PSP1群体190个F2个体的基因分型, 最终得到729个在190份F2群体单株中有清晰条带的多态性标记, 用于后续遗传连锁图谱的构建。然后利用这729个多态性标记对PSP2的亲本及随机选择的4个F2个体进行多态性检测, 最终在480个F2个体中成功筛选了103个多态性标记用于PSP2的遗传连锁图谱构建。
此外, 本研究还利用125个已知遗传连锁群位置信息的可公开获得的SSR标记[10,39-41]在2个群体中筛选锚定标记, 分别在PSP1和PSP2中鉴定出11个和17个锚定标记, 其中有3个标记为2个群体共有的锚定标记, 共计25个锚定标记, 这些标记可在后续遗传连锁图谱构建中用于分配连锁群(表1)。
2.2 遗传连锁图谱构建
获得所有样本的基因型数据后, 首先对所有标记进行数据完整性和偏分离检测。对于PSP1作图群体, 740个标记(包含11个锚定标记)中, 有43个标记缺失信息大于20%, 占总标记数的5.81%, 而有80个标记检测到偏分离现象, 占总标记数的10.81%。因此, 排除这123个标记后, 共有617个标记用于后续的遗传连锁图谱分析。针对PSP1构建的遗传连锁图谱, 将603个标记分配到7条连锁群上。根据11个锚定标记, PSP1图谱的7条连锁群分别对应于以往发表的遗传连锁图谱的6条连锁群[10,39-41], 有2条连锁群均对应于LGI, 而缺少对应于LGV的连锁群, 可能是由于缺乏LGV的锚定标记。此外, 该图谱全长5330.6 cM, 相邻标记之间的平均距离为8.9 cM。每条连锁群的长度从494.9 cM (LGII)到904.7 cM (LGIV)不等, 平均为761.5 cM; 每条连锁群的标记数目从62 (LGII)到113 (LGI-2)不等, 平均为86个标记(图1和表2)。
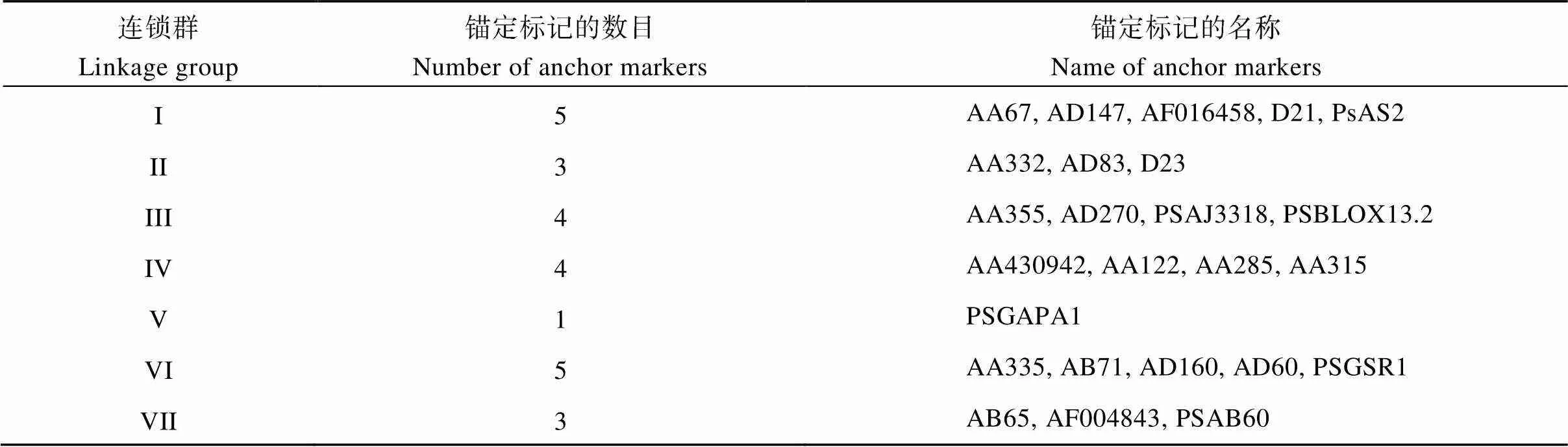
表1 筛选得到的25个锚定标记在以往发表豌豆遗传连锁图谱的分布

图1 豌豆PSP1群体遗传连锁图谱
粗体代表锚定标记, 标记名称以“e”开头的为EST-SSR标记。
The bold labels on the marker name represent anchor markers, and marker names started with “e” represent EST-SSR markers.
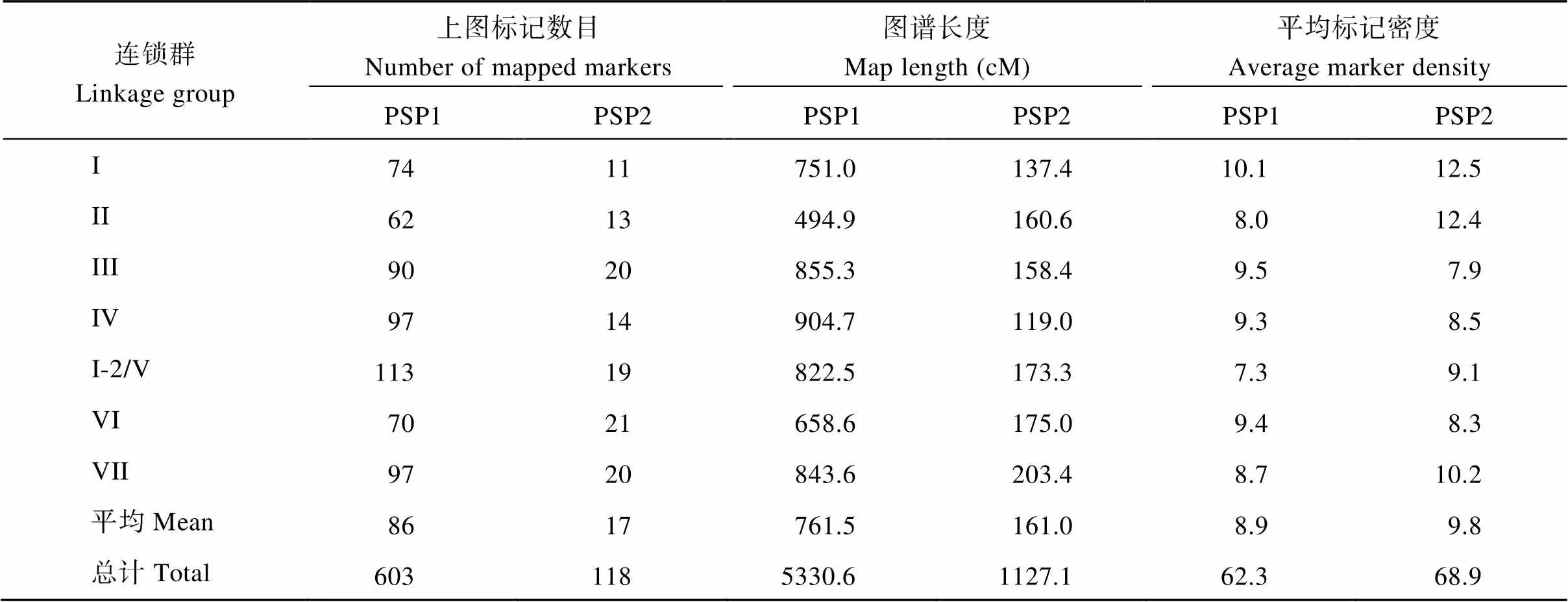
表2 利用豌豆PSP1和PSP2群体构建的2个遗传连锁图谱的标记分布
对于PSP2作图群体来说, 120个标记(包含17个锚定标记)中, 仅有1个标记由于缺失大于20%而被排除在后续分析之外。剩余的119个标记被用于进一步的连锁作图分析, 结果发现上图的118个标记分布在7条连锁群上。根据17个锚定标记, 这7条连锁群刚好完全对应于以往发表的遗传连锁图谱的7个连锁群[10,39-41]。此外, 该图谱的累积长度为1127.1 cM, 相邻标记之间的平均遗传距离为9.8 cM (图2和表2)。每条连锁群的长度从119.0 cM (LG IV)到203.4 cM (LGVII)不等, 平均为161.0 cM; 每条连锁群的标记数目从11 (LGI)到21 (LGVI)不等, 平均为17个标记(图2和表2)。
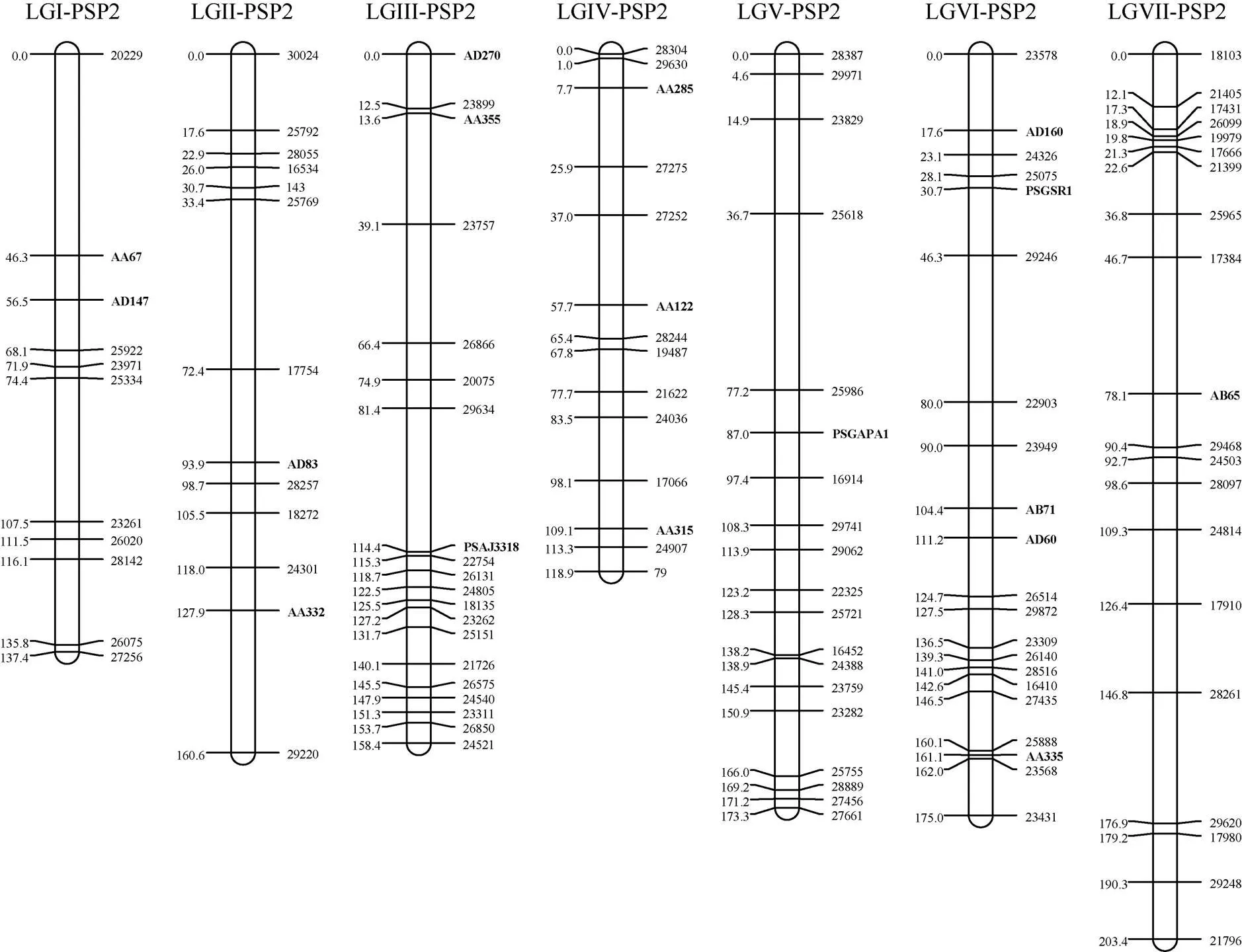
图2 豌豆PSP2群体遗传连锁图谱
粗体代表锚定标记。The bold labels on the marker name represent anchor markers.
2.3 整合遗传连锁图谱构建
基于以上2个遗传连锁图谱, 通过两两比较共发现了53个共有标记, 每个连锁群上的共有标记数为3 (LGI)到14 (LGVII)不等(表3)。利用上述2个遗传连锁图上的53个共有标记, 我们构建了一张包含668个SSR标记的整合遗传连锁图谱(标记信息详见附表1), 分布在7条连锁群上, 累积长度为6592.6 cM, 相邻标记之间的平均距离为10.0 cM。每条连锁群的长度从682.7 cM (LGII)到1077.2 cM (LGIII)不等, 平均为941.8 cM。在这7条连锁群中, 分布在LGV的标记数量最多, 有125个标记, 同时标记密度也最低, 为8.1 cM; 另有3条连锁群包含的标记数也都超过了100, 分别是LGIV (104)、LGVII (103)和LGIII (102); 而标记数量最少的连锁群为LGII, 仅有68个标记, 累积长度也是最小的, 仅有682.7 cM (图3和表3)。

图3 豌豆PSP1和PSP2整合遗传连锁图谱
粗体代表锚定标记, 下画线代表共有标记, 粗体加下画线代表共有锚定标记。标记名称以“e”开头的为EST-SSR标记。
The bold, underlined and bold underlined labels on the marker name represent anchor markers, common markers and common anchor markers, respectively. Marker names started with “e” represent EST-SSR markers.
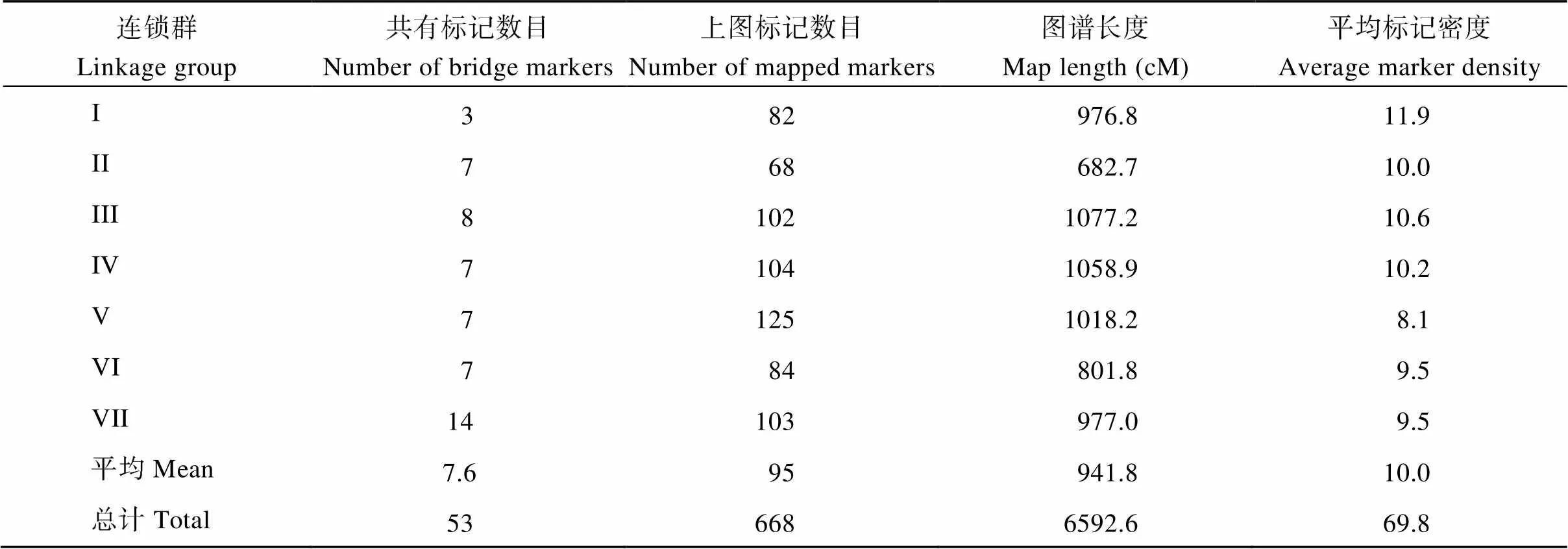
表3 豌豆整合遗传连锁图谱上的标记分布
将我们的整合图谱与以往的遗传图谱相比较, 结果发现, 本研究中的7条连锁群能够与以往遗传连锁图谱中7条连锁群相对应[10,42-44]。通过比较整合遗传图谱和2个单独的遗传图谱之间共有标记的顺序和位置, 结果发现, LGIV和LGVII这2个连锁群在不同图谱中的标记顺序存在较高的共线性, 而其他连锁群则由于作图群体差异而观察到了一些倒位和标记重排的现象。此外, 在整合图谱的LGV上, 同时包含2个锚定标记, 一个是在以往图谱中定位于LGV的PSGAPA1, 另一个是在以往图谱中定位于LGI的锚定标记AD147, 因而导致LGV的定义存疑(附图1)。
本研究以新近发表的豌豆基因组为参考(Caméor genome build 1a)[20], 对50个共有标记的扩增片段序列进行了BLAST比对分析, 从而对本文得到的遗传图谱和豌豆的物理图谱相比较(附表2)。发现有45个标记被成功比对到豌豆的7条染色体上, 上述有争议的1条连锁群(LGI-2/LGV)被证明为LGV。其中30个标记具有唯一比对位置, 15个标记比对到2个以上的位置; 剩下的5个标记比对到了未挂载到染色体的组装支架上。此外, 还比较了3个遗传图谱和物理图谱的标记顺序(附图1), 发现chr2LG1与LGI-PSP2高度一致, chr6LG2与LGII-PSP1高度一致, chr5LG3与LGIII-Integrated map基本一致, chr4LG4与LGIV- PSP1、LGIV-PSP2和LGIV-Integrated map基本一致, chr3LG5与LGV-PSP2和LGV-Integrated map基本一致, chr1LG6与LGVI-PSP2和LGVI-Integrated map基本一致, chr7LG7与LGIV-PSP1、LGIV-PSP2和LGIV-Integrated map一致率较高。
3 讨论
3.1 SSR标记筛选
由于具有多态性高、多等位基因、共显性、可重复、可转移和基因组覆盖度高等特性, SSR标记包括基因组SSR和基于表达序列标签的EST-SSR已被广泛应用于种质资源鉴定评价、遗传变异检测、遗传进化分析、遗传连锁图谱构建、功能基因定位以及标记辅助育种等诸多领域[21,48-50]。通过对基因组SSR和EST-SSR的应用范围进行比较发现, 基因组SSR因为多态性较高更适合于区分遗传关系比较近的基因型, 并且在指纹图谱构建和种质资源鉴定研究中更具优势; 而从基因或表达序列标签数据生成的EST-SSR标记则有利于获得更多基因的相关信息, 在重要农艺性状的标记辅助选择中具有更大优势, 同时在近缘物种之间具有更高的可转移性[21,51]。以往的研究已经成功开发了一些SSR标记[10-12,38,44], 并用于遗传多样性评估[1,33,52-55], 遗传连锁图谱构建[10,15], 以及标记性状关联分析[32,56]等。然而, 豌豆作为具有重要经济价值和显著生态优势的食用豆类作物之一, 目前具有明确定位信息同时可公开获取的基因组SSR和EST-SSR标记数量还相对有限, 这极大地阻碍了豌豆的分子遗传研究和标记辅助育种。在本研究中, 我们利用大规模筛选从本实验室开发的12,491个SSR标记中得到了729个多态性SSR标记, 同时从125个已发表的具有明确遗传图谱定位信息的标记中筛选了25个锚定标记, 分别用于2个基于中国豌豆种质F2群体的遗传连锁图谱构建和整合。在最终得到的整合遗传图谱中, 上图标记达到668个, 包括509个基因组SSR、134个EST-SSR和25个锚定标记, 分布在7条连锁群上, 对应于豌豆的7条染色体, 相关标记信息详见附表1。这些已定位的SSR标记将为豌豆种质资源鉴定、遗传关系分析、分子遗传作图以及标记辅助选择提供宝贵资源。
3.2 遗传连锁图谱构建
高密度遗传连锁图谱是进行遗传研究和分子育种的有力工具。针对豌豆的遗传连锁作图已经有很长的研究历史, 前人利用不同的分子标记和不同的作图群体已经构建了许多豌豆遗传连锁图谱[31]。随着二代测序技术的发展, 基因组SSR、EST-SSR和SNP标记的高通量开发为豌豆的分子作图奠定了重要基础[14-18,57]。最近, 有学者还利用SRAP、SSR和SNP这3种标记基于F2群体构建了一张豌豆的遗传连锁图谱, 包含128个遗传标记, 分布在9个连锁群上, 然后他们利用9个SSR标记作为锚定标记, 将其中的6个连锁群与以往发表的遗传连锁图谱中的连锁群对应起来[58]。尽管在豌豆中已经构建了超过50张的遗传连锁图谱, 然而目前国际上还没有基于SSR标记构建的标记数目达500个以上的图谱, 因此豌豆SSR遗传连锁图谱无论在标记数目和密度上均有待完善[10,15]。此外, 前人的研究已经证明中国豌豆种质资源具有独特的遗传背景[1,32-33], 然而基于中国种质的豌豆遗传连锁图谱很少。过去的一项研究基于中国种质构建的豌豆遗传连锁图谱总长1518 cM, 仅包含157个SSR标记, 标记间平均距离为9.7 cM, 而且分布在11条连锁群上, 与豌豆的单倍体染色体数并不一致(2= 2= 14)[15]。在本研究中, 我们首先基于与以往研究[15]相同的作图群体(PSP1), 筛选了大量多态性SSR标记, 对以往的图谱进行加密。加密后的图谱累计长度扩展到5330.6 cM, 标记数目增加到603个, 标记间平均距离缩小为8.9 cM, 所有标记分布在7个连锁群上(图1和表2), 其中6条与以往发表的豌豆遗传图谱一一对应。此外, 我们还利用一个新的大样本作图群体(PSP2)构建了一张新的豌豆遗传连锁图谱, 并通过17个锚定标记将总共118个标记定位在与以前发表的豌豆遗传图谱完全一致的7条连锁群上(图2和表2)[10]。本研究基于中国豌豆种质构建的2个作图群体代表了与国外豌豆群体具有显著差异的基因组背景, 得到了2个SSR遗传连锁图谱, 与以往的研究相比[15], PSP1图谱在标记数目和密度上具有明显提高, 而PSP2在连锁群装配方面则具有明显改善, 这些图谱将为中国豌豆的标记辅助育种提供有力工具。
3.3 整合遗传图谱
整合图谱凭借较高的标记数目和密度以及更完整的基因组覆盖范围等优势[34-35], 在许多作物包括豌豆中均有应用。过去几十年, 在豌豆遗传连锁作图悠久的研究历史中, 前人已经通过整合来自多个作图群体的信息而获得了很多复合遗传连锁图谱[10,16,31,36-37,42,59-61]。2015年, 法国科学家利用13.2K的基因芯片对12个豌豆RIL群体进行基因分型, 同时整合了以往图谱中的2277个标记, 构建了一个包含15,079个标记和7条连锁群的高密度一致性连锁图谱, 该图谱全长794.9 cM, 平均标记间距离0.24 cM, 为评估豌豆基因组结构、基因含量和进化历史以及候选基因鉴定提供了有力工具[16]。然而上述一致性图谱包含的EST-SSR和基因组SSR标记仅有187个, 为了在豌豆遗传连锁图谱上积累最大数量的SSR标记, 本研究通过53个共有标记将2个F2作图群体的数据合并构建了一个整合遗传连锁图谱, 覆盖范围为6592.6 cM, 包括668个SSR标记(509 基因组SSR、134 EST-SSR和25个锚定标记), 分布在7条连锁群上(图3和表3)。值得注意的是, 遗传连锁图谱的长度随标记数目的增多而延伸, 这种现象在包括豌豆在内的多个物种中均有发现, 有人推测可能与重组事件和偏分离有关[36-37,62-64]。此外, 有学者认为延伸的图谱长度对图谱上标记的映射顺序几乎没有影响[61]。过去的研究发现, 由于多等位标记、数据缺失、偏分离和染色体重排等原因, 在比较不同杂交群体获得的遗传连锁图谱时, 会出现标记定位和顺序的不一致[10,36-37,65]。在本研究中, 通过对2个单独的遗传连锁图谱之间53个共有标记的比较发现, 基于2个群体构建的遗传图谱均可组装到7条连锁群上, 与以往研究中的7个连锁群具有较好的对应关系, 然而PSP1图谱的LGI-2 (包含LGI锚定标记AD147)对应于PSP2图谱的LGV (包含LGV锚定标记PSGAPA1), 导致整合图谱中LGV的不确定性(附图1)。但是由于PSP1图谱中7个连锁群有6个与以往发表的图谱一一对应, 仅剩1个连锁群(LGI-2)因为缺乏额外的锚定标记无法与以往发表的LGV相对应; 同时PSP1图谱中的LGI-2与PSP2图谱中的LGV之间存在7个共有标记, 基于上述两点原因, 我们推测LGI-2应该对应于以往发表图谱中的LGV。然后通过对50个共有标记的扩增片段序列的BLAST比对分析, 对3个遗传图谱和物理图谱进行共线性比较, 结果支持有争议的一条连锁群确实对应于以往遗传图谱中的LGV (附表2), 而该连锁群上的锚定标记AD147可能是异位所致。另一方面, 单独图谱和整合图谱共线性比较发现, 不同图谱在LGIV和LGVII这2个连锁群上的标记顺序具有高度一致性, 而在其他连锁群上则观察到标记的颠倒和错位(附图1), 推测这种不一致性可能是由不同作图群体中发生的染色体重排引起的[10,36-37]。不同遗传图谱与物理图谱的共线性比较得到了类似的结果(附图1和附表2), 物理图谱的chr4LG4和chr7LG7与3个遗传图谱在LGIV和LGVII这2个连锁群上的标记顺序具有较高的一致性, 说明这2条连锁群或染色体在不同群体的亲本间并未发生明显结构变异; 物理图谱中的chr2LG1、chr3LG5和chr1LG6与PSP2遗传图谱的LGI、LGV和LGVI基本一致, 而与PSP1遗传图谱的标记顺序全部或部分相反, 说明PSP1群体的亲本在这3条连锁群或染色体上可能发生了倒位; 物理图谱中的chr6LG2与PSP1遗传图谱LGII的标记顺序基本一致, 而与PSP2遗传图谱的标记顺序全部相反, 说明PSP2群体的亲本在这条连锁群或染色体上可能发生了倒位;物理图谱中的chr5LG3与LGIII-Composite map的标记顺序一致, 而与PSP1和PSP2遗传图谱的标记顺序相反, 可能是参考基因组的豌豆基因型在该条染色体上发生了倒位或者错误组装。
4 结论
利用2个基于中国豌豆种质的F2群体, 通过QTL IciMapping V4.0软件, 整合得到一张包含7条连锁群、668个SSR标记、总遗传距离为6592.6 cM、平均标记密度为10 cM的豌豆遗传连锁图谱。本研究将为豌豆的分子遗传研究和标记辅助育种提供有力工具。
附表和附图 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuow xb.aspx。
[1] Zong X X, Redden R J, Liu Q C, Wang S M, Guan J P, Liu J, Xu Y H, Liu X J, Gu J, Yan L, Ades P, Ford R. Analysis of a diverse globalsp collection and comparison to a Chinese localcollection with microsatellite markers., 2009, 118: 193–204.
[2] Smýkal P, Aubert G, Burstin J, Coyne C J, Ellis N T H, Flavell A J, Ford R, Hýbl M, Macas J, Neumann P, McPhee K E, Redden R J, Rubiales D, Weller J L, Warkentin T D. Pea (L.) in the genomic era., 2012, 2: 74–115.
[3] Tian S J, Kyle W S A, Small D M. Pilot scale isolation of proteins from field peas (L.) for use as food ingredients., 1999, 34: 33–39.
[4] Santalla M, Amurrio J M, De Ron A M. Food and feed potential breeding value of green, dry and vegetable pea germplasm., 2001, 81: 601–610.
[5] FAOSTAT. [2020-03-14]. http://www.fao.org/faostat/en/#data.
[6] MacWilliam S, Wismer M, Kulshreshtha S. Life cycle and economic assessment of Western Canadian pulse systems: The inclusion of pulses in crop rotations., 2014, 123: 43–53.
[7] Semagn K, Bjornstad A, Ndjiondjop M N. Principles, requirements and prospects of genetic mapping in plants., 2006, 5: 2569–2587.
[8] Dirlewanger E, Isaac P G, Ranade S, Belajouza M, Cousin R, de Vienne D. Restriction fragment length polymorphism analysis of loci associated with disease resistance genes and developmental traits inL., 1994, 88: 17–27.
[9] Laucou V, Haurogne K, Ellis N , Rameau C. Genetic mapping in pea. 1. RAPD-based genetic linkage map of., 1998, 97: 905–915.
[10] Loridon K, McPhee K, Morin J, Dubreuil P, Pilet-Nayel M L, Aubert G, Rameau C, Baranger A, Coyne C, Lejeune-Henaut I, Burstin J. Microsatellite marker polymorphism and mapping in pea (L.)., 2005, 111: 1022–1031.
[11] DeCaire J, Coyne C J, Brumett S, Shultz J L. Additional pea EST-SSR markers for comparative mapping in pea (L.)., 2012, 131: 222–226.
[12] Mishra R K, Gangadhar B H, Nookaraju A, Kumar S, Park S W. Development of EST-derived SSR markers in pea () and their potential utility for genetic mapping and transferability., 2012, 131: 118–124.
[13] Leonforte A, Sudheesh S, Cogan N O I, Salisbury P A, Nicolas M E, Materne M, Forster J W, Kaur S. SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (L.)., 2013, 13: 161.
[14] Sindhu A, Ramsay L, Sanderson L A, Stonehouse R, Li R, Condie J, Shunmugam A S K, Liu Y, Jha A B, Diapari M, Burstin J, Aubert G, Tar’an B, Bett K E, Warkentin T D, Sharpe A G. Gene-based SNP discovery and genetic mapping in pea., 2014, 127: 2225–2241.
[15] Sun X L, Yang T, Hao J J, Zhang X Y, Ford R, Jiang J Y, Wang F, Guan J P, Zong X X. SSR genetic linkage map construction of pea (L.) based on Chinese native varieties., 2014, 2: 170–174.
[16] Tayeh N, Aluome C, Falque M, Jacquin F, Klein A, Chauveau A, Berard A, Houtin H, Rond C, Kreplak J, Boucherot K, Martin C, Baranger A, Pilet-Nayel M L, Warkentin T D, Brunel D, Marget P, Le Paslier M C, Aubert G, Burstin J. Development of two major resources for pea genomics: the GenoPea 13.2K SNP Array and a high-density, high-resolution consensus genetic map., 2015, 84: 1257–1273.
[17] Boutet G, Carvalho S A, Falque M, Peterlongo P, Lhuillier E, Bouchez O, Lavaud C, Pilet-Nayel M L, Riviere N, Baranger A. SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population., 2016, 17: 121.
[18] Ma Y, Coyne C J, Grusak M A, Mazourek M, Cheng P, Main D, McGee R J. Genome-wide SNP identification, linkage map construction and QTL mapping for seed mineral concentrations and contents in pea (L.)., 2017, 17: 43.
[19] Barilli E, Cobos M J, Carrillo E, Kilian A, Carling J , Rubiales D. A high-density integrated DArTseq SNP-based genetic map ofand identification of QTLs controlling rust resistance., 2018, 9: 167.
[20] Kreplak J, Madoui M A, Capal P, Novak P, Labadie K, Aubert G, Bayer P E, Gali K K, Syme R A, Main D, Klein A, Berard A, Vrbova I, Fournier C, d’Agata L, Belser C, Berrabah W, Toegelova H, Milec Z, Vrana J, Lee H, Kougbeadjo A, Terezol M, Huneau C, Turo C J, Mohellibi N, Neumann P, Falque M, Gallardo K, McGee R, Tar’an B, Bendahmane A, Aury J M, Batley J, Le Paslier M C, Ellis N, Warkentin T D, Coyne C J, Salse J, Edwards D, Lichtenzveig J, Macas J, Dolezel J, Wincker P, Burstin J. A reference genome for pea provides insight into legume genome evolution., 2019, 51: 1411–1422.
[21] Kalia R K, Rai M K, Kalia S, Singh R, Dhawan A K. Microsatellite markers: an overview of the recent progress in plants., 2011, 177: 309–334.
[22] Vieira M L C, Santini L, Diniz A L, Munhoz C D. Microsatellite markers: what they mean and why they are so useful., 2016, 39: 312–328.
[23] Ali A, Pan Y B, Wang Q N, Wang J D, Chen J L, Gao S J. Genetic diversity and population structure analysis ofandgenera using microsatellite (SSR) markers., 2019, 9: 10.
[24] Hao L, Zhang G S, Lu D Y, Hu J J, Jia H X. Analysis of the genetic diversity and population structure ofbased on phenotypic traits and simple sequence repeat markers., 2019, 7: e6419.
[25] Choi J K, Sa K J, Park D H, Lim S E, Ryu S H, Park J Y, Park K J, Rhee H I, Lee M, Lee J K. Construction of genetic linkage map and identification of QTLs related to agronomic traits in DH population of maize (L.) using SSR markers., 2019, 41: 667–678.
[26] Yang T, Jiang J Y, Zhang H Y, Liu R, Strelkov S, Hwang S F, Chang K F, Yang F, Miao Y M, He Y H, Zong X X. Density enhancement of a faba bean genetic linkage map () based on simple sequence repeats markers., 2019, 138: 207–215.
[27] Anjani K, Ponukumatla B, Mishra D, Ravulapalli D P. Identification of simple-sequence-repeat markers linked to Fusarium wilt (f. sp) resistance and marker- assisted selection for wilt resistance in safflower (L.) interspecific offsprings., 2018, 137: 895–902.
[28] Swathi G, Rani C V D, Md J, Madhav M S, Vanisree S, Anuradha C, Kumar N R, Kumar N A P, Kumari K A, Bhogadhi C, Ramprasad E, Sravanthi P, Raju S K, Bhuvaneswari V, Rajan C P D, Jagadeeswar R. Marker-assisted introgression of the major bacterial blight resistance genes,and, and blast resistance gene,, into the popular rice variety, JGL1798., 2019, 39: 12.
[29] Kumar N, Shikha D, Kumari S, Choudhary B K, Kumar L, Singh I S. SSR-based DNA Fingerprinting and diversity assessment among Indian germplasm of: an aquatic underutilized and neglected food crop., 2018, 185: 34–41.
[30] Siew G Y, Ng W L, Tan S W, Alitheen N B, Tan S G, Yeap S K. Genetic variation and DNA fingerprinting of durian types in Malaysia using simple sequence repeat (SSR) markers., 2018, 6: e4266.
[31] Tayeh N, Aubert G, Pilet-Nayel M L, Lejeune-Henaut I, Warkentin T D, Burstin J. Genomic tools in pea breeding programs: Status and perspectives., 2015, 6: 1037.
[32] Liu R, Fang L, Yang T, Zhang X Y, Hu J G, Zhang H Y, Han W L, Hua Z K, Hao J J, Zong X X. Marker-trait association analysis of frost tolerance of 672 worldwide pea (L.) collections., 2017, 7: 5919.
[33] Wu X B, Li N N, Hao J J, Hu J G, Zhang X Y, Blair M W. Genetic diversity of Chinese and global pea (L.) collections., 2017, 57: 1–11.
[34] Milczarski P, Bolibok-Brągoszewska H, Myśków B, Stojałowski S, Heller-Uszyńska K, Góralska M, Brągoszewski P, Uszyński G, Kilian A, Rakoczy-Trojanowska M. A high density consensus map of rye (L.) based on DArT markers., 2011, 6: e28495.
[35] Blenda A, Fang D D, Rami J F, Garsmeur O, Luo F, Lacape J M. A high density consensus genetic map of tetraploid cotton that integrates multiple component maps through molecular marker redundancy check., 2012, 7: e45739.
[36] Sudheesh S, Lombardi M, Leonforte A, Cogan N O I, Materne M, Forster J W, Kaur S. Consensus genetic map construction for field pea (L.), trait dissection of biotic and abiotic stress tolerance and development of a diagnostic marker for thepowdery mildew resistance gene., 2015, 33: 1391–1403.
[37] Sudheesh S, Rodda M, Kennedy P, Verma P, Leonforte A, Cogan N O I, Materne M, Forster J W, Kaur S. Construction of an integrated linkage map and trait dissection for bacterial blight resistance in field pea (L.)., 2015, 35: 185.
[38] Yang T, Fang L, Zhang X Y, Hu J G, Bao S Y, Hao J J, Li L, He Y H, Jiang J Y, Wang F, Tian S, Zong X X. High-throughput development of SSR markers from pea (L.) based on next generation sequencing of a purified Chinese commercial variety., 2015, 10: e0139775.
[39] Kwon S J, Brown A F, Hu J, McGee R, Watt C, Kisha T, Timmerman-Vaughan G, Grusak M, McPhee K E, Coyne C J. Genetic diversity, population structure and genome-wide marker-trait association analysis emphasizing seed nutrients of the USDA pea (L.) core collection., 2012, 34: 305–320.
[40] Kaur S J, Pembleton L W, Cogan N O, Savin K W, Leonforte T, Paull J, Materne M, Forster J W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers., 2012, 13: 104.
[41] Xu S C, Gong Y M, Mao W H, Hu Q Z, Zhang G W, Fu W, Xian Q Q. Development and characterization of 41 novel EST-SSR markers for(Leguminosae)., 2012, 99: E149–E153.
[42] Bordat A, Savois V, Nicolas M, Salse J, Chauveau A, Bourgeois M, Potier J, Houtin H, Rond C, Murat F, Marget P, Aubert G, Burstin J. Translational genomics in legumes allowed placing in silico 5460 unigenes on the pea functional map and identified candidate genes inL.2011, 1: 93–103.
[43] 顾竟, 李玲, 宗绪晓, 王海飞, 关建平, 杨涛. 豌豆种质表型性状SSR标记关联分析. 植物遗传资源学报, 2011, 12: 833–839. Gu J, Li L, Zong X X, Wang H F, Guan J P, Yang T. Association analysis between morphological traits of pea and its polymorphic SSR markers., 2011,12: 833–839 (in Chinese with English abstract).
[44] Burstin J, Deniot G, Potier J, Weinachter C, Aubert G, Barranger A. Microsatellite polymorphism in., 2001, 120: 311.
[45] Dellaporta S L, Wood J, Hicks J B. A plant DNA minipreparation: version II., 1983, 1: 19–21.
[46] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations., 2015, 3: 269–283.
[47] Voorrips R E. MapChart: software for the graphical presentation of linkage maps and QTLs., 2002, 93: 77–78.
[48] Parida S K, Kalia S K, Kaul S, Dalal V, Hemaprabha G, Selvi A, Pandit A, Singh A, Gaikwad K, Sharma T R, Srivastava P S, Singh N K, Mohapatra T. Informative genomic microsatellite markers for efficient genotyping applications in sugarcane., 2009, 118: 327–338.
[49] Kamaluddin, Khan M A, Kiran U, Ali A, Abdin M Z, Zargar M Y, Ahmad S, Sofi P A, Gulzar S. Molecular markers and marker- assisted selection in crop plants. In: Abdin M Z, Kiran U, Kamaluddin, Ali A, eds. Plant Biotechnology: Principles and Applications. Singapore: Springer Singapore, 2017. pp 295–328.
[50] Nadeem M A, Nawaz M A, Shahid M Q, Doğan Y, Comertpay G, Yıldız M, Hatipoğlu R, Ahmad F, Alsaleh A, Labhane N, Özkan H, Chung G, Baloch F S. DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing., 2018, 32: 261–285.
[51] Varshney R K, Graner A, Sorrells M E. Genic microsatellite markers in plants: features and applications., 2005, 23: 48–55.
[52] Smýkal P, Hybl M, Corander J, Jarkovsky J, Flavell A J, Griga M. Genetic diversity and population structure of pea (L.) varieties derived from combined retrotransposon, microsatellite and morphological marker analysis., 2008, 117: 413–424.
[53] 宗绪晓, 关建平, 王述民, 刘庆昌. 中国豌豆地方品种SSR标记遗传多样性分析. 作物学报, 2008, 34: 1330–1338. Zong X X, Guan J P, Wang S M, Liu Q C. Genetic diversity among Chinese pea (L.) landraces revealed by SSR markers., 2008, 34: 1330–1338 (in Chinese with English abstract).
[54] Zong X X, Ford R, Redden R R, Guan J P, Wang S M. Identification and analysis of genetic diversity structure withinbased on microsatellite markers., 2009, 8: 257–267.
[55] 宗绪晓, 关建平, 王述民, 刘庆昌, Redden R R, Ford R. 国外栽培豌豆遗传多样性分析及核心种质构建. 作物学报, 2008, 34: 1518–1528. Zong X X, Guan J P, Wang S M, Liu Q C, Redden R R, Ford R. Genetic diversity and core collection of alienL. germplasm., 2008, 34: 1518–1528 (in Chinese with English abstract).
[56] Prakash N, Kumar R, Choudhary V K, Singh C M. Molecular assessment of genetic divergence in pea genotypes using microsatellite markers., 2016, 39: 183–188.
[57] Duarte J, Riviere N, Baranger A, Aubert G, Burstin J, Cornet L, Lavaud C, Lejeune-Henaut I, Martinant J P, Pichon J P, Pilet-Nayel M L, Boutet G. Transcriptome sequencing for high throughput SNP development and genetic mapping in pea., 2014, 15: 126.
[58] Guindon M F, Martin E, Cravero V, Gali K K, Warkentin T D, Cointry E. Linkage map development by GBS, SSR, and SRAP techniques and yield-related QTLs in pea., 2019, 39: 54.
[59] Aubert G, Morin J, Jacquin F, Loridon K, Quillet M C, Petit A, Rameau C, Lejeune-Henaut I, Huguet T, Burstin J. Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume., 2006, 112: 1024–1041.
[60] Duarte J, Riviere N, Baranger A, Aubert G, Burstin J, Cornet L, Lavaud C, Lejeune-Henaut I, Martinant J P, Pichon J P, Pilet-Nayel M L, Boutet G. Transcriptome sequencing for high throughput SNP development and genetic mapping in pea., 2014, 15: 126.
[61] Sindhu A, Ramsay L, Sanderson L A, Stonehouse R, Li R, Condie J, Shunmugam A S K, Liu Y, Jha A B, Diapari M, Burstin J, Aubert G, Tar’an B, Bett K E, Warkentin T D, Sharpe A G. Gene-based SNP discovery and genetic mapping in pea., 2014, 127: 2225–2241.
[62] Sybenga J. Recombination and chiasmata: few but intriguing discrepancies., 1996, 39: 473–484.
[63] Knox M R, Ellis T H N. Excess heterozygosity contributes to genetic map expansion in pea recombinant inbred populations., 2002, 162: 861–873.
[64] Truong S K, McCormick R F, Morishige D T, Mullet J E. Resolution of genetic map expansion caused by excess heterozygosity in plant recombinant inbred populations., 2014, 4: 1963–1969.
[65] Ellis T H, Turner L, Hellens R P, Lee D, Harker C L, Enard C, Domoney C, Davies D R. Linkage maps in pea., 1992, 130: 649–663.
An integrated high-density SSR genetic linkage map from two F2population in Chinese pea
LIU Rong1,**, WANG Fang1,**, FANG Li1,**, YANG Tao1, ZHANG Hong-Yan1, HUANG Yu-Ning1, WANG Dong1,3, JI Yi-Shan1, XU Dong-Xu2, LI Guan1, GUO Rui-Jun1, and ZONG Xu-Xiao1,*
1Center for Crop Germplasm Resources, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;2Institute of Legumes Crop, Zhangjiakou Academy of Agricultural Sciences, Zhangjiakou 075000, Hebei, China;3Shandong Center of Crop Germplasm Resources, Jinan 250100, Shandong, China
Pea (L.) is an important food legume crop grown widely throughout the world for humans or livestock consumption. Genetic linkage map constructed with SSR markers have played a vital role in marker-assisted breeding of many crops including pea. Public available and transferable SSR markers and genetic linkage map with sufficient SSR markers based on genetically distinct Chinese pea germplasm are limited despite a long study history on genetic linkage mapping in pea. In this study, in order to obtain more transferable SSR markers and high resolution genetic linkage maps for Chinese pea, 617 polymorphic SSR markers were firstly screened from 12,491 genome-wide SSR markers and some related literatures by our laboratory, and these SSR markers were used to construct an enhanced genetic linkage map for the G0003973 × G0005527 F2population. The enhanced genetic linkage map covered 5330.6 cM in total length containing 603 SSR markers with an average inter-marker distance of 8.8 cM, which was significantly improved both in marker number and in density compared with the previous map. 119 polymorphic SSR markers were screened based on the above results to develop a new map for a large W6-22600 × W6-15174 F2population including 118 SSR markers with a cumulative length of 1127.1 cM assembled into seven genetic linkage groups. Furthermore, data from the above two genetic maps were combined to build an integrated map of 6592.6 cM, comprising 668 SSR markers, 509 genomic SSRs, 134 EST-SSRs and 25 anchor markers distributed in seven genetic linkage groups. These SSR markers and genetic linkage maps will provide a valuable tool for the genetic study and marker-assisted breeding in pea.
pea (L.); SSR; genetic linkage map; integrated map; marker-assisted breeding
10.3724/SP.J.1006.2020.04028
本研究由国家自然科学基金项目(31801428), 国家现代农业产业技术体系建设专项(CARS-08), 农作物种质资源保护与利用专项(2019NWB036-07), 中国农业科学院科技创新工程, 国家农作物种质资源共享服务平台(NICGR2019)和国家重点研发计划项目(2017YFE0105100)资助。
This study was supported by the National Natural Science Foundation of China (31801428), the China Agriculture Research System (CARS-08), the Crop Germplasm Resources Protection (2019NWB036-07), the Agricultural Science and Technology Innovation Program (ASTIP) in CAAS, the National Infrastructure for Crop Germplasm Resources Project from the Ministry of Science and Technology of China (NICGR2019), and the National Key Research and Development Program of China (2017YFE0105100).
宗绪晓, E-mail: zongxuxiao@caas.cn
**同等贡献(Contributed equally to this work)
刘荣, E-mail: liurong@caas.cn; 王芳, E-mail: fwang11@huskers.unl.edu;方俐, E-mail: hathor_fang@163.com
2020-02-08;
2020-04-15;
2020-05-09.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20200509.1330.002.html
猜你喜欢
廉政瞭望·下半月(2022年4期)2022-05-12
中学数学杂志(初中版)(2021年3期)2021-08-09
北华大学学报(自然科学版)(2021年3期)2021-07-13
今日农业(2021年2期)2021-03-19
集装箱化(2019年7期)2019-10-18
中学数学杂志(初中版)(2019年4期)2019-09-18
课外生活(小学1-3年级)(2017年5期)2017-06-10
中学生数理化·中考版(2016年7期)2016-12-07
故事作文·高年级(2016年9期)2016-10-25
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
