
用全基因组关联分析筛选甘蓝型油菜叶片叶绿素含量候选基因
2020-09-14 07:22:32荐红举高玉敏李阳阳魏丽娟刘列钊李加纳
作物学报 2020年10期
荐红举 霍 强 高玉敏 李阳阳 谢 玲 魏丽娟 刘列钊 卢 坤 李加纳,*
用全基因组关联分析筛选甘蓝型油菜叶片叶绿素含量候选基因
荐红举1,2,**霍 强1,2,**高玉敏1,2李阳阳1,2谢 玲1,2魏丽娟1,2刘列钊1,2卢 坤1,2李加纳1,2,*
1西南大学农学与生物科技学院, 重庆 400715;2西南大学现代农业科学研究院, 重庆 400715
提高油菜产量是保障国家粮油安全的重要举措。作物“源”“流”“库”理论表明, 充足的光合产物(源)是高产的前提, 而叶绿素是直接参与光合作用的物质, 因此, 选育高叶绿素含量的甘蓝型油菜是提高产量的重要途径。本课题组前期对全球收集的588份优异油菜种质资源进行5X重测序, 获得385,692个高质量SNP标记。利用SPAD-502叶绿素仪于2018—2019连续2年测定苗期完全伸展的叶片叶绿素总量, 结合获得的SNP标记进行全基因组关联分析(genome-wide association study, GWAS), 筛选与叶绿素含量显著关联的SNP位点。结果表明, 2018年鉴定到5个显著关联的SNP位点, 贡献率为5.51%~7.89%, 其中S6_3493805位点贡献率最大; 2019年检测到46个SNP位点, 贡献率为7.29%~10.34%, 其中S13_11413088位点贡献率最大。将显著关联SNP位点上下游各500 kb区间内的基因与参考基因组比对, 初步筛选出2022个油菜基因。将其基因序列在拟南芥基因组内进行BLAST比对, 结合前人已报道的拟南芥同源基因功能, 筛选到23个候选基因, 其中5个属于叶绿素合成途径同源基因。本研究结果为后续油菜叶片叶绿素含量的遗传改良奠定了基础。
甘蓝型油菜; 叶绿素含量; 全基因组关联分析; 候选基因
油菜是中国四大油料作物之一, 也是产油效率最高的油料作物, 而菜籽作为我国食用植物油的最大来源, 自给率严重不足, 因此, 提高油菜产量和含油量是油菜育种的迫切目标。叶绿素是光合作用的重要色素, 筛选并研究控制油菜叶绿素含量的基因对提高油菜产量具有重要意义。
叶绿素在光合作用中能够吸收光能并参与能量传递[1]。在生物学和遗传学上已经对叶绿素代谢进行了广泛的研究[2-4]。叶绿素的生物合成首先是叶绿素前体L-谷氨酰tRNA (ALA)到原卟啉IX的合成[4-5], 然后通过ALA脱水酶(ALAD)将ALA的2个分子缩合成吡咯分子, 即胆色素原。再通过酶的连续转化和原卟啉IX的金属螯合反应来划分通路, 从而指导最终产物叶绿素和血红素的形成[4]。在黑暗中, 叶绿素的生物合成分支在中间原叶绿素酸酯(Pchlide)被阻断, 因为原叶绿素酸酯向叶绿素的转化是由依赖光的NADPH催化的[6]。然而, 过量的原叶绿素酸酯游离和其他吡啶中间体在黑暗中积累可能在光照射下产生活性氧(ROS), 从而导致子叶光漂白甚至细胞死亡[7]。因此, 叶绿素的生物合成在植物生长发育过程中扮演着重要的角色。叶绿素生物合成相关基因的表达差异造成了叶绿素含量的不同。
叶片是植物光合作用的主要场所, 也是植物源到库的重要源场所。而植物叶色突变体的叶色主要表现在苗期。甘蓝型油菜中的叶色黄化突变体BnaC.ygl是由于血红素加氧酶(HO1)的活性降低导致血红素的积累, 从而抑制了叶绿素合成前体ALA的合成, 造成叶绿素生物合成受到抑制导致叶色变黄[8]; 玉米中5个黄化突变体是由于、、、和缺失所引起, 这5个基因与磷酸烯醇式丙酮酸(PEP)合成相关, 相关基因的缺失导致细胞中含有较少的质体核糖体和光合复合物, 表明PEP在叶绿素生物合成中起到关键作用[9]。植物光合作用可以通过叶绿素将CO2和H2O转化为可储存的有机物, 因此叶绿素是植物有机物积累的重要物质。通过降低叶绿素降解的时间可以增加产量[10]。另外, 叶片中叶绿素的含量也与作物粒饱满程度、有机物积累及单株产量显著相关[11]。
近年来在高粱[12]、玉米[13]、小麦[14]、水稻[15]、大豆[16]等植物中进行了叶绿素相关研究, 但关于甘蓝型油菜叶片叶绿素含量相关基因鉴定的报道较少[17-18]。Ge等[19]利用白菜一个F2:3群体2年的叶绿素a和叶绿素b含量数据进行QTL定位, 一共检测到10个QTL, 单个QTL解释表型率为7%~17%。Wang等[17]利用一个甘蓝型油菜叶绿素缺失突变体和中双11构建的F2群体及620个F2:3群体将基因定位在一个长度为311 kb区间内, 该定位区间包括22个候选基因; Qian等[18]探究了甘蓝型油菜中的缺失对光系统(LHC)相关基因表达及光合反应中心蛋白PSI和PSII表达的影响以及对籽粒油脂积累的影响, 同时也发现携带基因也会影响株高。
随着高通量测序技术的成熟以及基因型分型成本的降低, 全基因组关联分析(genome-wide association)已经成为植物基因探究以及复杂性状解析的重要方法[20]。全基因组关联分析已经在棉花[21-22]、杨树[23]、大豆[24]等植物中广泛应用。本研究对588份甘蓝型油菜自然群体的叶片进行2年叶绿素含量测定并进行全基因组关联分析, 筛选出控制甘蓝型油菜叶片叶绿素含量相关的候选基因, 为油菜叶绿素含量的遗传改良奠定基础。
1 材料方法
1.1 试验材料
用于关联分析的588份甘蓝型油菜是由全球各地征集的油菜品种组成, 其中大部分来自中国重庆、湖北、湖南、江苏、陕西等地, 部分来自加拿大、德国、瑞典、丹麦、澳大利亚等国家。所有材料由重庆市西南大学油菜工程中心提供。
1.2 田间试验与叶绿素测定
该群体于2017—2018年, 2018—2019年连续种植于重庆市北碚区歇马镇油菜基地。采用随机区组设计, 2个重复, 以育苗移栽方式每小区种植3行, 每行15株, 行距40 cm, 株距20 cm。按照常规生产方式进行田间管理。在植株苗期用SPAD-502Plus叶绿素仪测定完全伸展的叶片中间部位叶绿素含量, 对每个品种测定5株。利用IBM SPSS Statistics 19.0统计分析软件进行描述性统计分析并制作正态分布图。
1.3 全基因组关联分析的方法
参考文献进行基因型数据测定与分析、群体结构、亲缘关系分析与连锁不平衡分析[25]。全基因组关联分析采用naive、Q、PCA、K、Q+K和PCA+K六种统计模型来评估群体结构和亲缘关系的影响(Q: 群体结构; PCA: 主成分; K: 亲缘关系), 进行表型和SNP关联分析。运用TASSEL 5.0软件进行这6种模型的关联分析, 其中使用一般线性模型(GLM)分析naive、Q和PCA模型, 使用混合线性模型(MLM)分析K、Q+K和PCA+K模型。利用SAS软件检测这6种模型的运算结果, 对其lg()的观测值和lg()期望值绘制Quantile-quantile散点图, 通过QQ图比较后确定最佳模型, 在最佳模型下进行叶片叶绿素含量的全基因组关联分析, 利用R软件作Manhattan图。本试验使用的SNP数据为385,692个,值小于阈值(1/385,692 = 2.593E-06)的位点为显著关联位点。
1.4 候选基因的挖掘
根据甘蓝型油菜“Darmor-bzh”参考基因组[26], 提取分析得到的叶片叶绿素含量显著关联的SNP标记各位点500 kb内的基因[27]; 将需要功能分析的基因序列与拟南芥所有基因序列进行BLASTn比对, e-value阈值为1e-10, 并以同源性最高的拟南芥基因作为待分析基因进行功能注释来筛选与叶绿素含量相关的候选基因。
1.5 候选基因验证
对叶绿素含量极端材料在苗期取3个生物学重复的叶片并提取RNA。根据前期所取材料的叶片提取的RNA, 使用TaKaRa公司的PrimeScript RT reagent Kit with gDNA Eraser反转录合成cDNA, 用于qRT-PCR验证。以油菜作为内参基因, 使用TaKaRa公司的SYBR Premix ExII试剂盒进行qRT-PCR验证候选基因的相对表达量。设置每个样品3次技术重复。在BIO-RAD定量PCR仪上进行qRT-PCR扩增。在网站http://biodb.swu.edu.cn/ qprimerdb上查找设计候选基因的qRT-PCR引物(表1), 由上海生工生物工程有限公司合成。
2 结果与分析
2.1 叶绿素含量表型分析
自然群体中, 叶绿素含量在2018年和2019年的变异系数分别为11.27%和12.73%, 2年间的变异系数较为稳定(表2)。自然群体2年叶绿素含量表现出连续正态分布(图1), 说明该性状符合由多基因控制的数量性状特点, 适合全基因组关联分析。
表1 候选基因引物
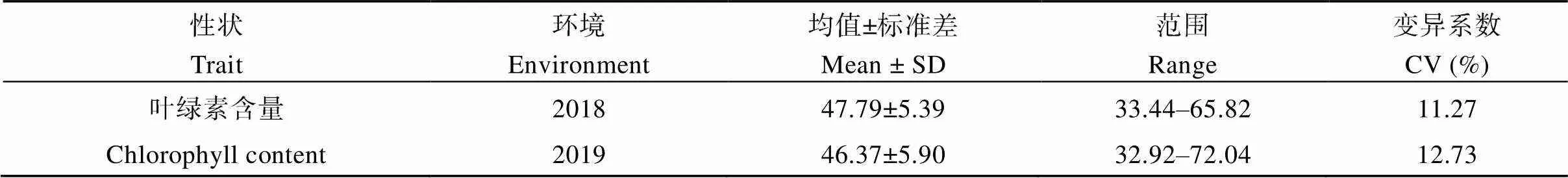
表2 甘蓝型油菜叶绿素含量的表型数据统计分析
SD: standard deviation; CV: coefficient of variation.
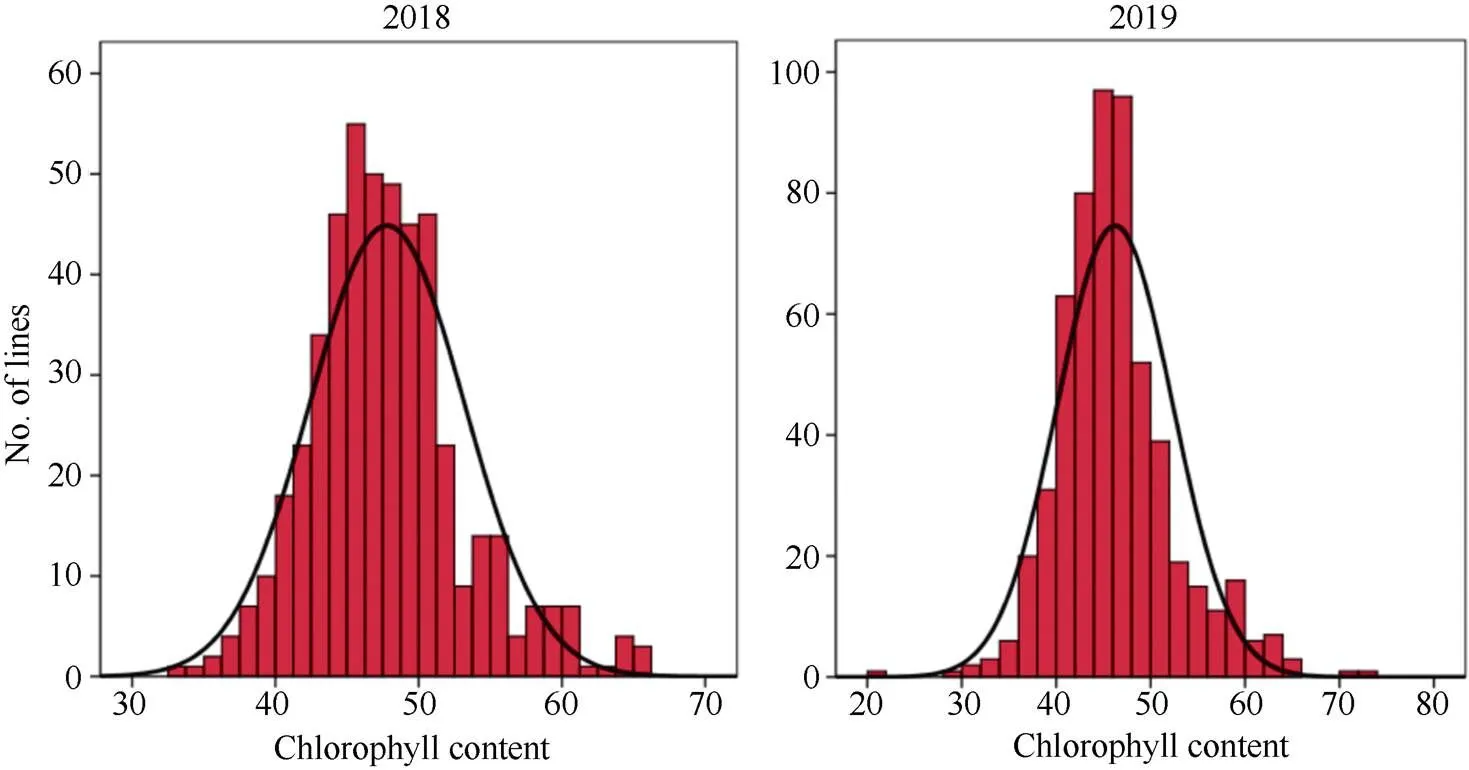
图1 甘蓝型油菜叶片叶绿素含量的频次分布
2.2 甘蓝型油菜叶片叶绿素含量全基因组关联分析
对叶绿素含量关联分析6种模型下的结果绘制Quantile-quantile散点图(图2), 由图2可知, 最佳模型为P+K模型。在最佳模型下, 利用385,692个SNPs, 以值小于阈值(1/385,692 = 2.593E-06)确定显著关联SNP位点(表3), 并绘制Manhattan图(图3)。
2018年检测到5个SNP位点, 分别位于A01 (1个)、A06 (2个)、A10 (1个)和C04 (1个)染色体, 它们的贡献率为5.51%~7.89%, 其中S6_3493805位点贡献率最大; 2019年检测到46个SNP位点, 分别位于A01 (8个)、A02 (1个)、A03 (1个)、A04 (1个)、A08 (5个)、A09 (1个)、A10 (1个)、C02 (2个)、C03 (8个)、C04 (4个)、C05 (1个)、C06 (10个)、C08 (2个)和C09 (1个)染色体, 贡献率为7.29%~10.34%, 其中S13_11413088位点贡献率最大。
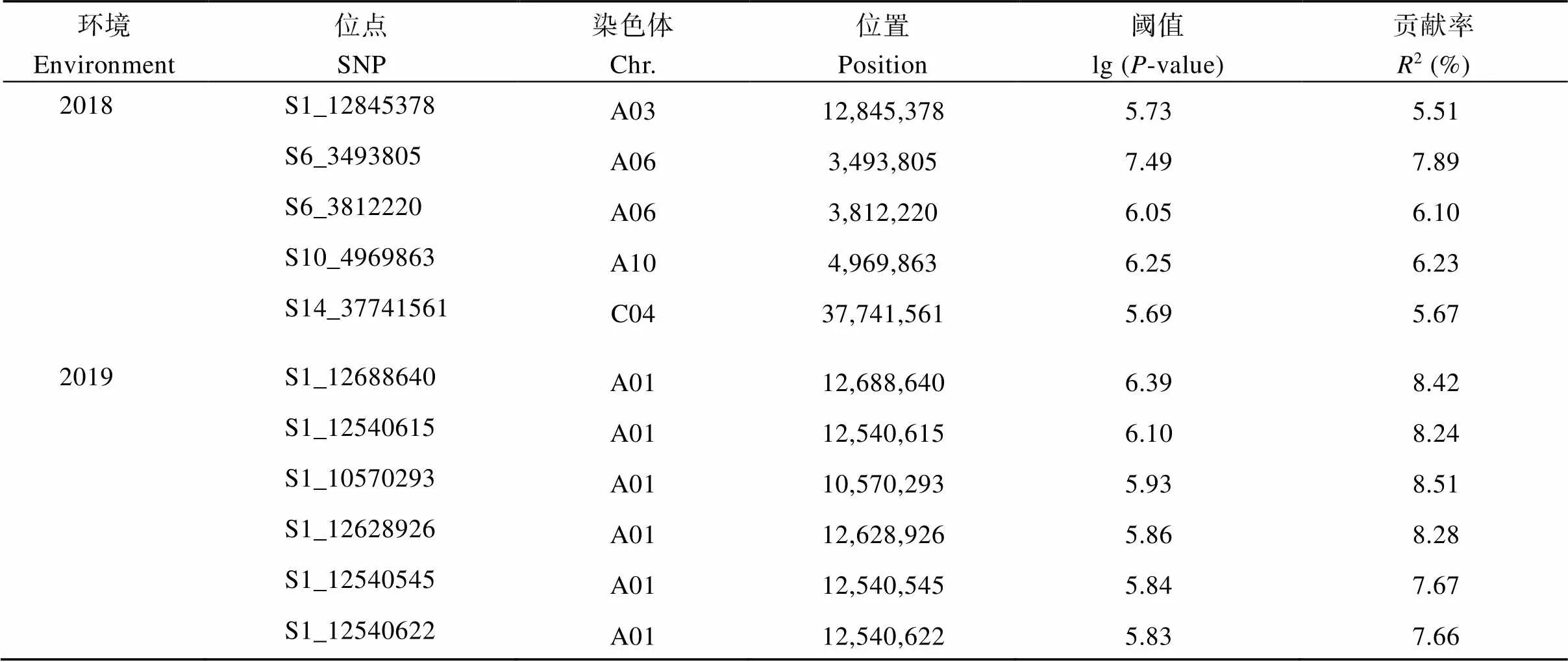
表3 最佳模型下叶绿素含量显著位点表
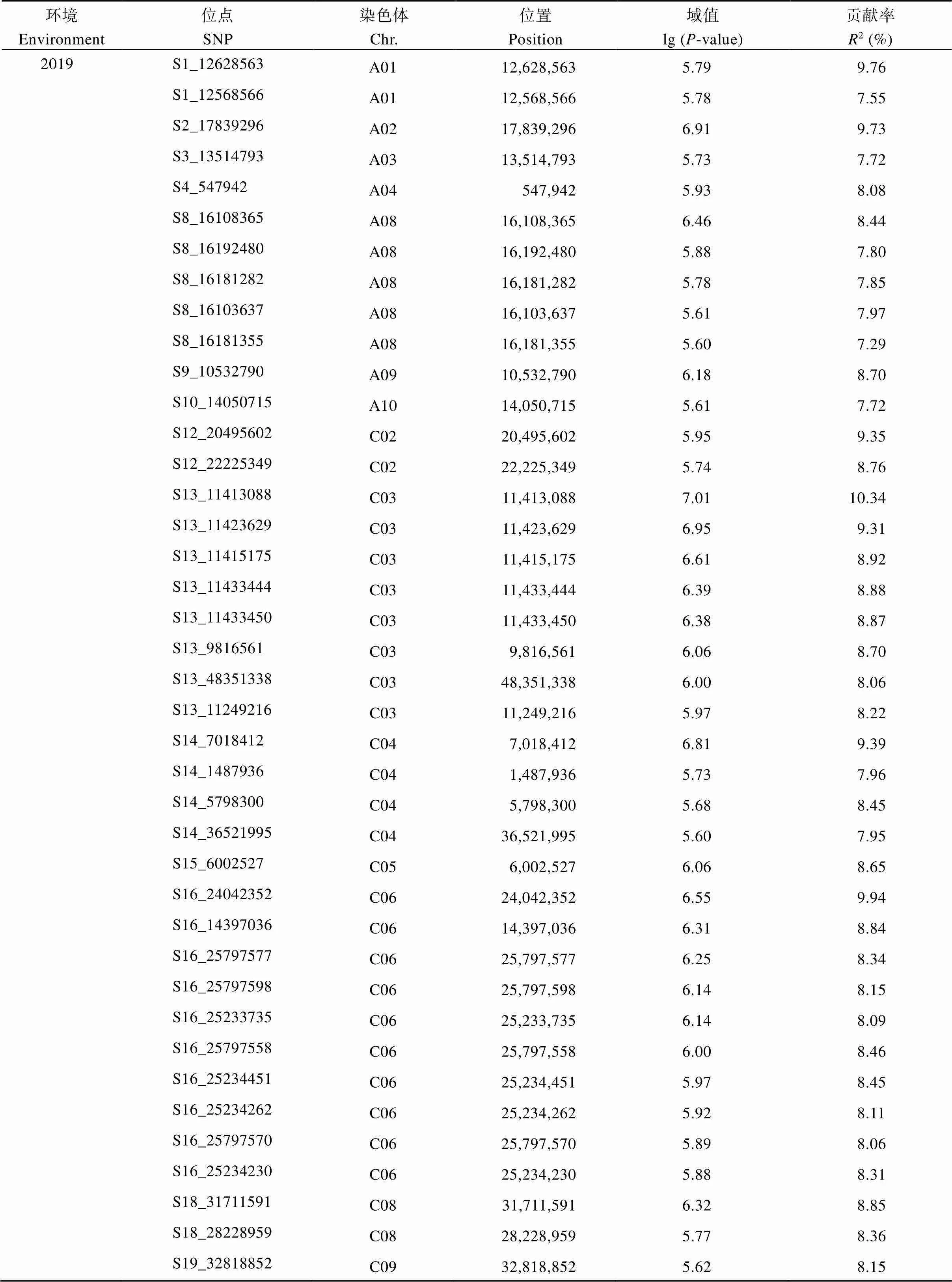
(续表3)
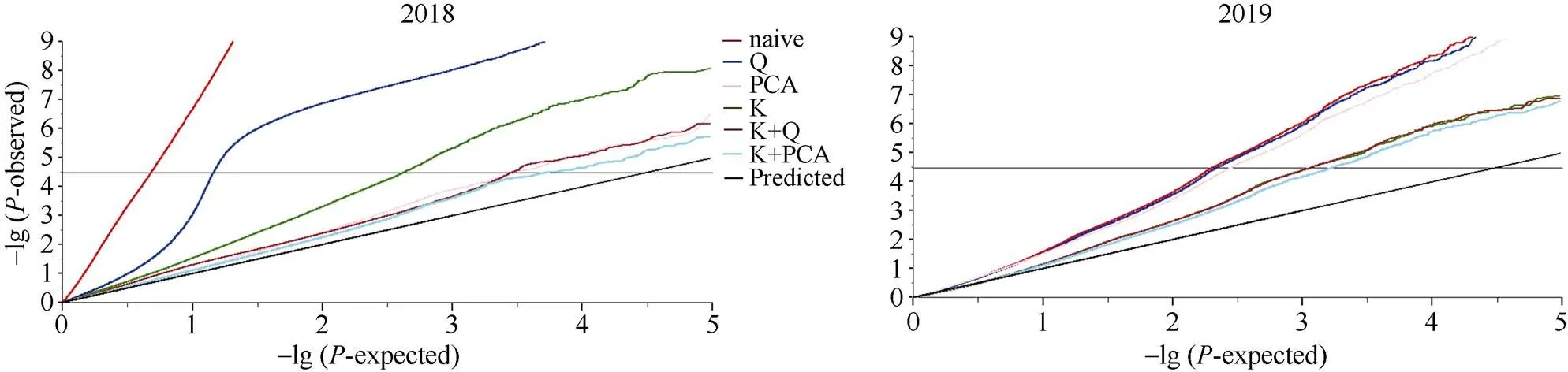
图2 甘蓝型油菜叶绿素含量在各模型中的QQ图
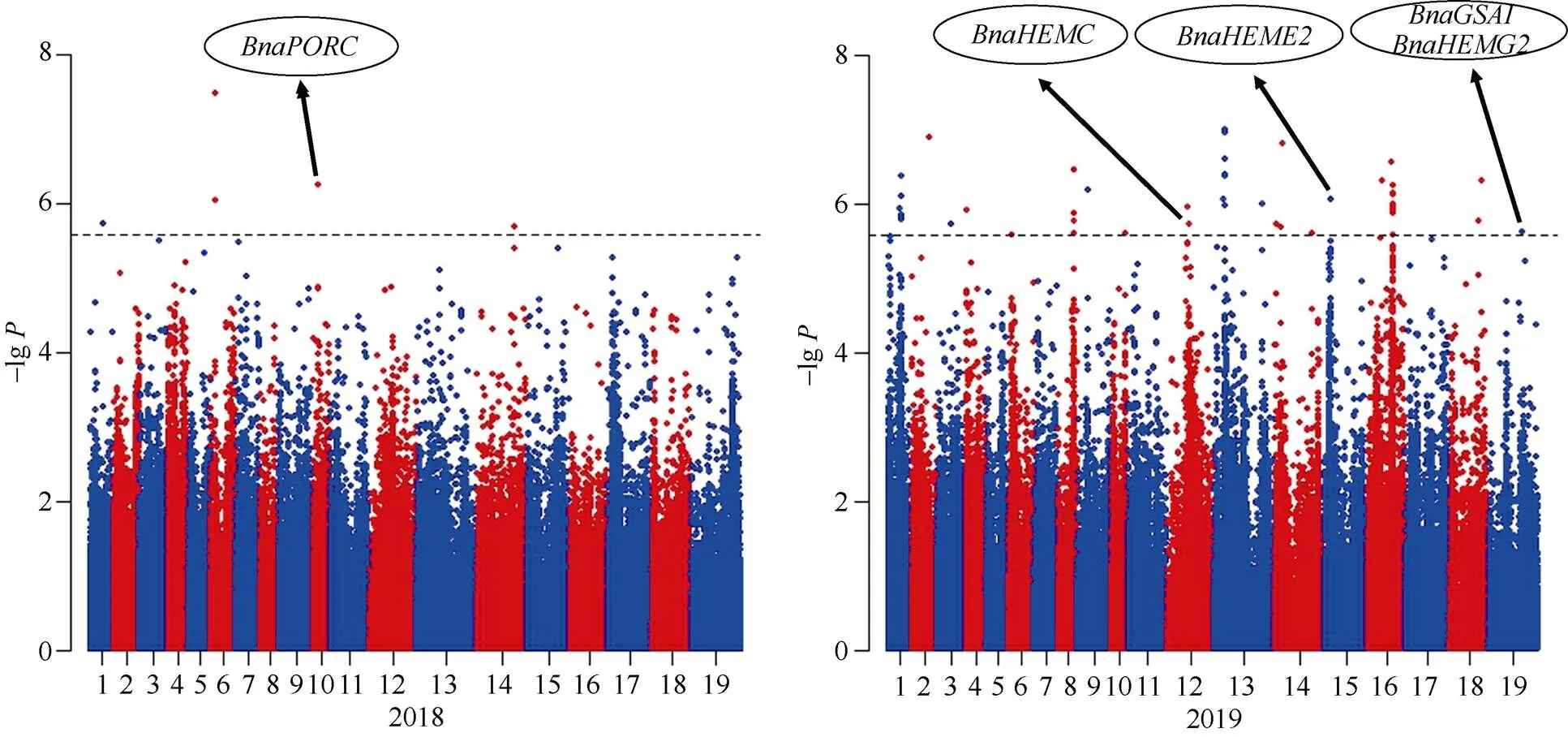
图3 甘蓝型油菜叶绿素含量在最佳模型中的曼哈顿图
2.3 甘蓝型油菜叶片叶绿素含量候选基因筛选
初步筛选出23个与油菜叶片叶绿素含量显著关联的候选基因, 与拟南芥基因序列进行Blastn比对, 结合前人已报道的拟南芥同源基因功能, 筛选可能在本研究中发挥作用的候选基因(表4)。
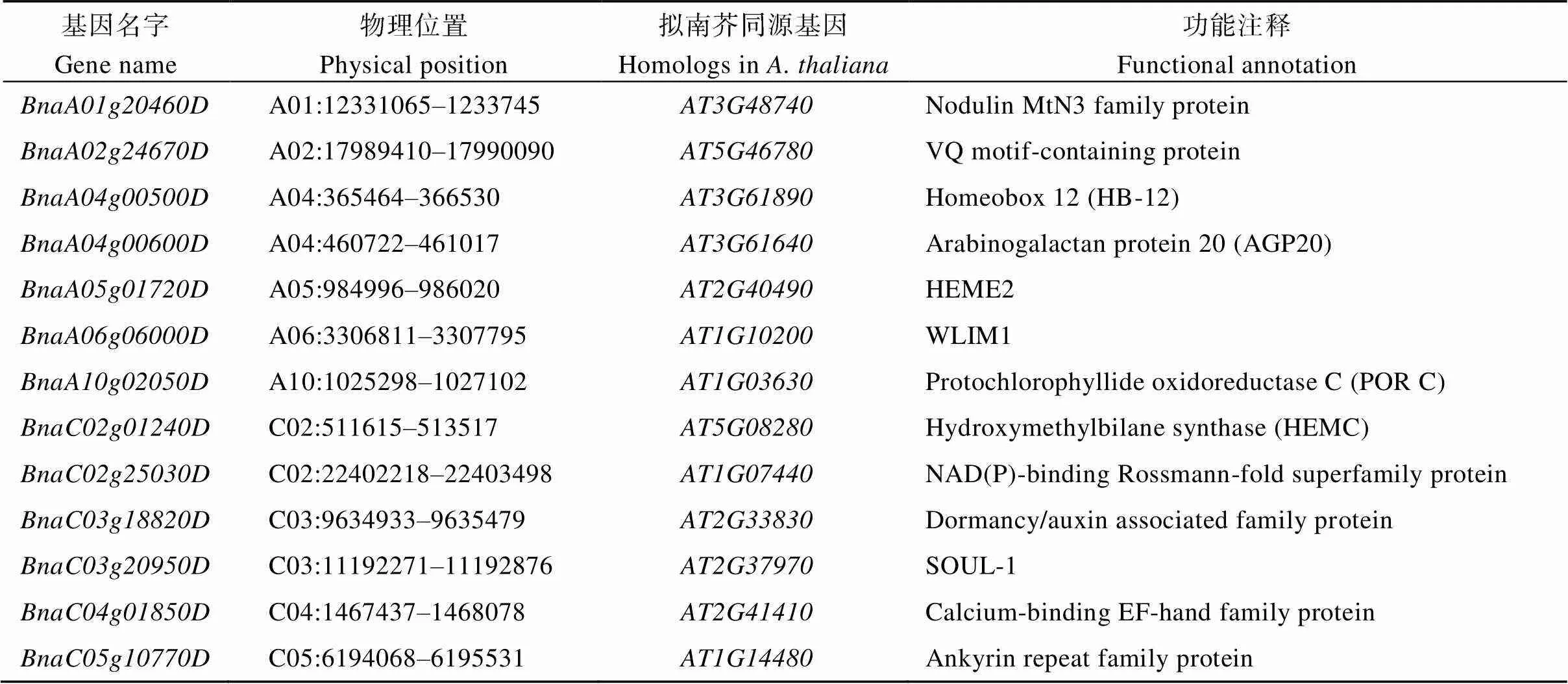
表4 叶绿素含量候选基因
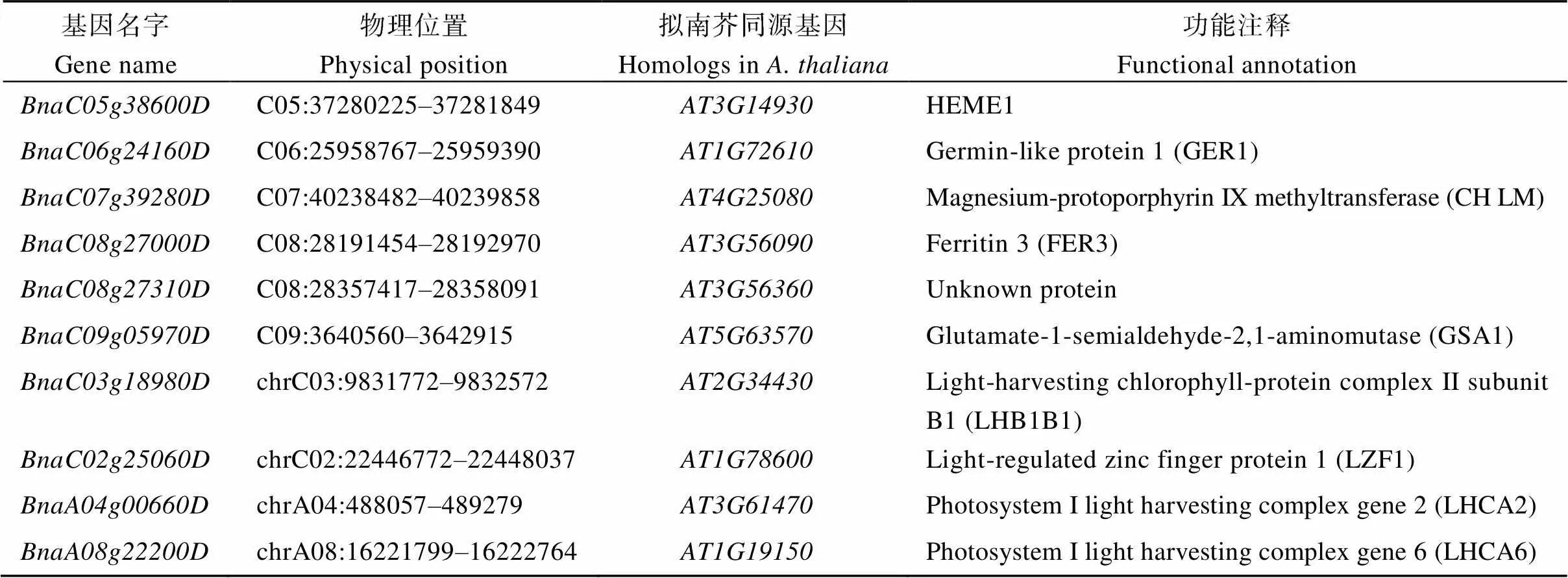
(续表4)
2.4 候选基因的验证
挑选20个候选基因进行荧光定量验证, 检测其在2类材料中的表达情况表明, 相比叶绿素含量高的材料, 在叶绿素含量低的材料中, 11个基因上调表达, 9个基因下调表达(图4)。上调基因包括, 其编码HEME1蛋白, 下调基因包括, 其编码HEMG2, 这些基因均参与叶绿素合成途径。
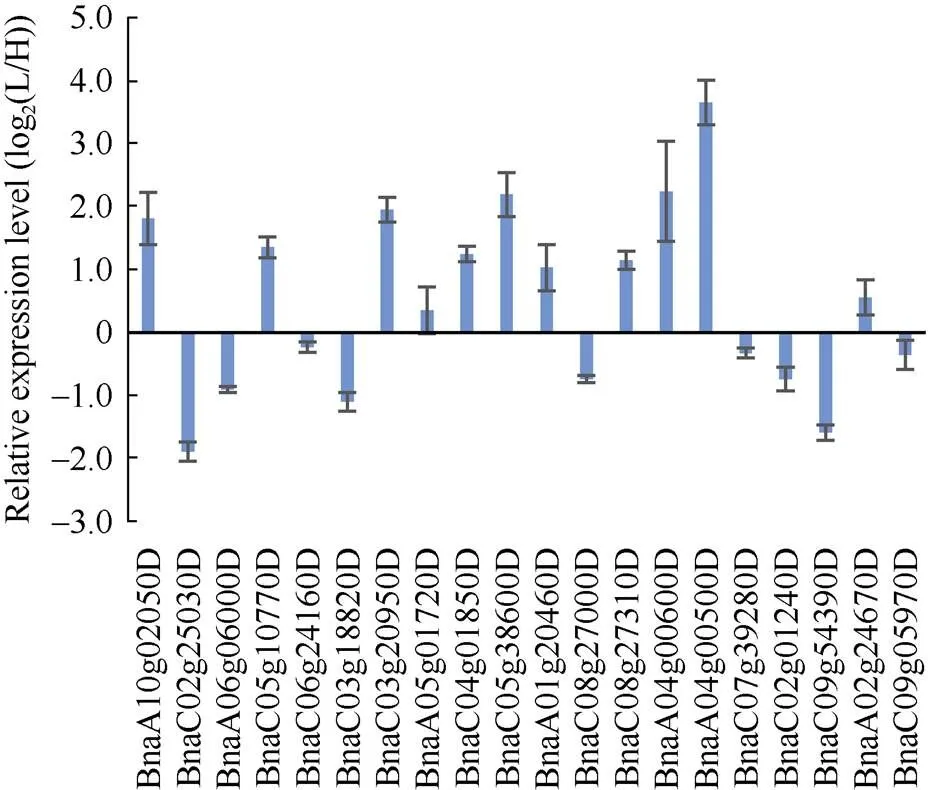
图4 甘蓝型油菜叶片叶绿素含量候选基因定量验证
3 讨论
利用数量性状座位(quantitative trait loci, QTL)或者全基因组关联分析可以快速鉴定与性状关联的分子标记和基因, 是非常有效的定位候选基因的方法。定位甘蓝型油菜重要农艺性状QTL已经在产量性状如千粒重[27]、角果长度[28]和农艺性状如开花期[29]、株高[30]等方面取得重要进展。利用GWAS定位甘蓝型油菜复杂的数量性状同样取得重要进展[31-33]。但是, 影响甘蓝型油菜叶片叶绿素含量相关QTL或者候选基因的研究相对较少, 仅有1篇通过突变体鉴定到的一个候选基因BnaC.YGL[8]。
本研究中, 利用588份全球搜集的优质种质资源, 具有较为丰富的表型变异, 叶绿素含量变异范围广(表2); 课题组前期已经对该资源材料进行重测序, 挖掘出385,692个高质量的SNP位点[25], 这些为通过GWAS分析筛选叶绿素含量显著关联位点奠定基础。植物合成的叶绿素含量不仅受到遗传因素的调控, 也会受到外界环境的影响, 如外界光照、温度和营养[34]。而当植物叶绿素含量发生变化, 细胞内叶绿素的超微结构、叶片生理生化特征以及叶色调控机制也都会发生改变[17]。油菜叶片叶绿素含量受年度间水肥条件和光温条件差异影响, 基因型与环境互作较大, 不同环境下有不同的基因群发挥作用, 进而造成关联位点的差异。通过测定甘蓝型油菜苗期叶片叶绿素含量, 2018和2019年分别筛选出与叶绿素显著关联的SNP位点5个和46个。通过对SNP位点上下游各500 kb范围内候选基因的筛选,共得到2022个候选基因, 其中,()、()、()和() 4个基因的同源基因在拟南芥中参与叶绿素合成途径(表4)。这些将作为重要候选基因进一步分析。
另外, 4个基因参与叶绿体发育和光合作用过程,如, 编码叶绿素a/b结合蛋白(LIGHT-HARVESTING CHLOROPHYLL-PROTEIN COMPLEX II SUBUNIT B1, LHB1B1), 该蛋白定位在叶绿体膜、类囊体膜上参与光捕获, 响应光刺激[35]。和分别编码光合系统I的光捕获复合体基因2 (PHOTOSYSTEM I LIGHT HARVESTING COMP LEX GENE 2, LHCA2)和LHCA6, 都定位在叶绿体类囊体膜, 参与光捕获过程[36-37]。编码光调控的锌指蛋白1 (light-regulated zinc finger protein 1, LZF1), 又名B-box domain protein 22 (BBX22和DBB3), 其定位在细胞核, 参与含花青素的复合生物合成过程、叶绿素生物合成过程、叶绿体组织等调控过程[38-39]。
4 结论
2年分别鉴定到5个和46个显著关联的SNP标记, SNP位点上下游500 kb区间内共筛选出23个与叶绿素合成途径相关的候选基因, 并进行了定量验证, 本研究为油菜后续叶片叶绿素含量的遗传改良奠定了基础。
[1] Von Wettstein D, Gough S, Kannangara C G. Chlorophyll biosynthesis., 1995, 7: 1039–1057.
[2] Eckhardt U, Grimm B, Hortensteiner S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants., 2004, 56: 1–14.
[3] Tanaka A, Tanaka R. Chlorophyll metabolism., 2006, 9: 248–255.
[4] Tanaka R, Tanaka A. Tetrapyrrole biosynthesis in higher plants., 2007, 58: 321–346.
[5] Mochizuki N, Tanaka R, Grimm B, Masuda T, Moulin M, Smith A G, Tanaka A, Terry M J. The cell biology of tetrapyrroles: a life and death struggle., 2010, 15: 488–498.
[6] Solymosi K, Schoefs B. Etioplast and etio-chloroplast formation under natural conditions: the dark side of chlorophyll biosynthesis in angiosperms., 2010, 105: 143–166.
[7] Buhr F, El Bakkouri M, Valdez O, Pollmann S, Lebedev N, Reinbothe S, Reinbothe C. Photoprotective role of NADPH: protochlorophyllide oxidoreductase A., 2008, 105: 12629–12634.
[8] Zhu L, Yang Z, Zeng X, Gao J, Liu J, Yi B, Ma C, Shen J, Tu J, Fu T, Wen J. Heme oxygenase 1 defects lead to reduced chlorophyll in., 2017, 93: 579–592.
[9] Williams-Carrier R, Zoschke R, Belcher S, Pfalz J, Barkan A. A major role for the plastid-encoded RNA polymerase complex in the expression of plastid transfer RNAs., 2014, 164: 239–248.
[10] Zhang K, Zhang Y, Chen G, Tian J. Genetic analysis of grain yield and leaf chlorophyll content in common wheat., 2009, 37: 499–511.
[11] Feng B, Liu P, Li G, Dong S T, Wang F H, Kong L A, Zhang J W. Effect of heat stress on the photosynthetic characteristics in flag leaves at the grain-filling stage of different heat-resistant winter wheat varieties., 2014, 200: 143–155.
[12] Kassahun B, Bidinger F R, Hash C T, Kuruvinashetti M S. Stay-green expression in early generation sorghum [(L.) Moench] QTL introgression lines., 2010, 172: 351–362.
[13] Cai H G, Chu Q, Yuan L X, Liu J C, Chen X H, Chen F J, Mi G H, Zhang F S. Identification of quantitative trait loci for leaf area and chlorophyll content in maize () under low nitrogen and low phosphorus supply., 2012, 30: 251–266.
[14] Czyczylo-Mysza I, Tyrka M, Marcinska I, Skrzypek E, Karbarz M, Dziurka M, Hura T, Dziurka K, Quarrie S A. Quantitative trait loci for leaf chlorophyll fluorescence parameters, chlorophyll and carotenoid contents in relation to biomass and yield in bread wheat and their chromosome deletion bin assignments., 2013, 32: 189–210.
[15] Jiang S, Zhang X, Zhang F, Xu Z, Chen W, Li Y. Identification and fine mapping of, a quantitative trait loci controlling the chlorophyll content from tillering to heading in rice (L.)., 2012, 103: 720–726.
[16] Dhanapal A P, Ray J D, Singh S K, Hoyos-Villegas V, Smith J R, Purcell L C, Fritschi F B. Genome-wide association mapping of soybean chlorophyll traits based on canopy spectral reflectance and leaf extracts., 2016, 16: 174.
[17] Wang Y K, He Y J, Yang M, He J B, Xu P, Shao M Q, Chu P, Guan R Z. Fine mapping of a dominant gene conferring chlorophyll-deficiency in., 2016, 6: 31419. doi: 10.1038/srep31419.
[18] Qian L W, Voss-Fels K, Cui Y X, Jan H U, Samans B, Obermeier C, Qian W, Snowdon R J. Deletion of a stay-green gene associates with adaptive selection in., 2016, 9: 1559–1569.
[19] Ge Y, Wang T, Wang N, Wang Z, Liang C, Ramchiary N, Choi S R, Lim Y P, Piao Z Y. Genetic mapping and localization of quantitative trait loci for chlorophyll content in Chinese cabbage (ssp.)., 2012, 147: 42–48.
[20] Luo J. Metabolite-based genome-wide association studies in plants., 2015, 24: 31–38.
[21] Su J J, Li L B, Zhang C, Wang C X, Gu L J, Wang H T, Wei H L, Liu Q B, Huang L, Yu S X. Genome-wide association study identified genetic variations and candidate genes for plant architecture component traits in Chinese upland cotton., 2018, 131: 1299–1314.
[22] Li T G, Ma X F, Li N Y, Zhou L, Liu Z, Han H Y, Gui Y J, Bao Y M, Chen J Y, Dai X F. Genome-wide association study discovered candidate genes of Verticillium wilt resistance in upland cotton (L.)., 2017, 15: 1520–1532.
[23] Fahrenkrog A M, Neves L G, Resende M F R, Vazquez A I, de los Campos G, Dervinis C, Sykes R, Davis M, Davenport R, Barbazuk W B, Kirst M. Genome-wide association study reveals putative regulators of bioenergy traits in., 2017, 213: 799–811.
[24] Wei W, Mesquita A C O, Figueiro A D, Wu X, Manjunatha S, Wickland D P, Hudson M E, Juliatti F C, Clough S J. Genome-wide association mapping of resistance to a Brazilian isolate ofin soybean genotypes mostly from Brazil., 2017, 18: 849.
[25] Lu K, Wei L J, Li X L, Wang Y T, Wu J, Liu M, Zhang C, Chen Z Y, Xiao Z C, Jian H J, Cheng F, Zhang K, Du H, Cheng X C, Qu C M, Qian W, Liu L Z, Wang R, Zou Q Y, Ying J M, Xu X F, Mei J Q, Liang Y, Chai Y R, Tang Z L, Wan H F, Ni Y, He Y J, Lin N, Fan Y H, Sun W, Li N N, Zhou G, Zheng H K, Wang X W, Paterson A H, Li J N. Whole-genome resequencing revealsorigin and genetic loci involved in its improvement., 2019, 10: 1154.
[26] Chalhoub B, Denoeud F, Liu S Y, Parkin I A P, Tang H B, Wang X Y, Chiquet J, Belcram H, Tong C B, Samans B. Early allopolyploid evolution in the post-Neolithicoilseed genome., 2014, 345: 950–953.
[27] Fu Y, Wei D Y, Dong H L, He Y J, Cui Y X, Mei J Q, Wan H F, Li J, Snowdon R, Friedt W, Li X R, Qian W. Comparative quantitative trait loci for silique length and seed weight in., 2015, 5: 14407.
[28] Wang X D, Chen L, Wang A N, Wang H, Tian J H, Zhao X P, Chao H B, Zhao Y J, Zhao W G, Xiang J, Gan J P, Li M T. Quantitative trait loci analysis and genome-wide comparison for silique related traits in., 2016, 16: 71.
[29] Jian H J, Zhang A X, Ma J Q, Wang T Y, Yang B, Shuang L S, Liu M, Li J N, Xu X F, Paterson A H, Liu L Z. Joint QTL mapping and transcriptome sequencing analysis reveal candidate flowering time genes inL., 2019, 9: 390.
[30] Shen Y S, Xiang Y, Xu E S, Ge X H, Li Z Y. Major co-localized QTL for plant height, branch initiation height, stem diameter, and flowering time in an alien introgression derivedDH population., 2018, 9: 390.
[31] Lu K, Xiao Z C, Jian H J, Peng L, Qu C M, Fu M L, He B, Tie L M, Liang Y, Xu X F, Li J N. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in., 2016, 6: 36452.
[32] Wang J, Xian X H, Xu X F, Qu C M, Lu K, Li J N, Liu L Z. Genome-wide association mapping of seed coat color in., 2017, 65: 5229–5237.
[33] Xiao Z C, Zhang C, Tang F, Yang B, Zhang L Y, Liu J S, Huo Q, Wang S F, Li S T, Wei L J, Du H, Qu C M, Lu K, Li J N, Li N N. Identification of candidate genes controlling oil content by combination of genome-wide association and transcriptome analysis in the oilseed crop., 2019, 12: 216.
[34] Ferreira V D S, Anna C S. Impact of culture conditions on the chlorophyll content of microalgae for biotechnological applications., 2017, 33: 20.
[35] Peltier J B, Ytterberg A J, Sun Q, van Wijk K J. New functions of the thylakoid membrane proteome ofrevealed by a simple, fast, and versatile fractionation strategy., 2004, 279: 49367–49383.
[36] Wientjes E, Croce R. The light-harvesting complexes of higher-plant Photosystem I: Lhca1/4 and Lhca2/3 form two red-emitting heterodimers., 2011, 433: 477–485.
[37] Otani T, Yamamoto H, Shikanai T. Stromal loop of lhca6 is responsible for the linker function required for the NDH-PSI supercomplex formation., 2017, 58: 851–861.
[38] Chang C S J, Li Y H, Chen L T, Chen W C, Hsieh W P, Shin J, Jane W N, Chou S J, Choi G, Hu J M, Somerville S, Wu S H. LZF1, a HY5-regulated transcriptional factor, functions inde-etiolation., 2008, 54: 205–219.
[39] Chang C S J, Maloof J N, Wu S H. COP1-mediated degradation of BBX22/LZF1 optimizes seedling development in., 2011, 156: 228–239.
Selection of candidate genes for chlorophyll content in leaves ofusing genome-wide association analysis
JIAN Hong-Ju1,2,**, HUO Qiang1,2,**, GAO Yu-Min1,2, LI Yang-Yang1,2, XIE Ling1,2, WEI Li-Juan1,2, LIULie-Zhao1,2, LU Kun1,2, and LI Jia-Na1,2,*
1College of Agronomy and Biotechnology, Southwest University, Chongqing 400715, China;2Academy of Agricultural Sciences, Southwest University, Chongqing 400715, China
Increasing rapeseed production is important to ensure the national food and oil security. According to the theory of crop source and sink, sufficient photosynthate (sources) is the premise of high yield, and chlorophyll is the important substance for photosynthesis. Therefore, breeding high chlorophyll contentis an important way to ensure high yield. In our previous study, 588 excellent germplasm resources collected worldwide were re-sequenced with 5and 385,692 high-quality SNP markers were obtained. SPAD-502 chlorophyll meter was used to measure the total chlorophyll content of mature leaves in 2018–2019. Genome wide association study (GWAS) was conducted to screen SNP sites significantly related to chlorophyll content. Five SNP loci were identified in 2018, with a contribution rate of 5.51%–7.89%, of which S6_3493805 had the largest contribution. 46 SNP loci were detected in 2019, with a contribution rate of 7.29%–10.34%, of which S13_11413088 had the largest contribution. In total, 2022 rapeseed genes were screened out by comparing the reference genome with genes in the regions of the 500 kb before and after the SNP. Based on the function ofhomologous genes previously reported, screened 23 candidate genes, among which five were homologous genes in chlorophyll synthesis pathway. These results lay a foundation for the genetic improvement of chlorophyll content in leaves ofin future.
; chlorophyll content; GWAS; candidate genes
10.3724/SP.J.1006.2020.04007
本研究由国家重点研发计划项目(2018YFD0100504-05)和重庆市民生工程主题专项(cstc2016shms-ztzx80020)资助。
This study was supported by the National Key Research and Development Program of China (2018YFD0100504-05) and the Special Project of Chongqing People’s Livelihood (cstc2016shms-ztzx80020).
李加纳, E-mail: ljn1950@swu.edu.cn, Tel: 023-68250701
**同等贡献(Contributed equally to this work)
荐红举, E-mail: hjjian518@swu.edu.cn; 霍强, E-mail: 354011524@qq.com
2020-01-12;
2020-04-15;
2020-04-27.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20200427.0825.004.html
猜你喜欢
今日农业(2021年21期)2021-11-26 05:07:00
今日农业(2021年14期)2021-10-14 08:35:40
阅读(科学探秘)(2020年8期)2020-11-06 06:22:48
西藏农业科技(2019年3期)2019-11-04 00:35:14
心声歌刊(2019年4期)2019-09-18 01:15:28
西藏农业科技(2019年1期)2019-07-25 00:37:02
中国果业信息(2019年1期)2019-01-05 17:41:42
西藏农业科技(2018年4期)2018-04-25 06:39:28
数学小灵通(1-2年级)(2017年10期)2017-11-08 08:39:47
生物学教学(2017年9期)2017-08-20 13:22:32
