
儿童瞬时受体电位阳离子通道蛋白6基因突变相关激素耐药型肾病综合征三例分析
2020-09-12 00:58史卓高春林夏正坤孙涛张沛
临床肾脏病杂志 2020年8期
史卓 高春林 夏正坤 孙涛 张沛
210002 南京,东部战区总医院儿科
原发性肾病综合征中大约有20%的患儿出现激素耐药,其中局灶节段硬化(focal segmental glomerulosclerosis,FSGS)是其主要的病理改变,且多发生于6岁前,并在10岁内进展为ESRD[1-2]。目前已明确多种基因的突变参与FSGS的形成,尤其是编码足细胞相关蛋白的单基因突变在其中发挥重要作用[3]。
瞬时受体电位阳离子通道6蛋白基因(TRPC6)编码的TPRC6蛋白[4]与足细胞裂孔隔膜(slit diaphragm,SD)的结构密切相关,共同参与足细胞间的信号传导和稳定结构等生理功能[5]。TRPC6基因突变会激活TRPC6蛋白的Ca2+通道,引起Ca2+内流增加,破坏足细胞的细胞骨架并进一步地引起肾小球的滤过功能异常,使得尿液中出现蛋白质,并最终进展成 FSGS[6]。
糖皮质激素类虽是肾病综合征的一线治疗用药,但对于FGSG患者,单用激素往往无法使患儿病情得到有效控制,需要与免疫抑制剂联合使用[7]。KDIGO诊疗指南特别推荐使用钙调磷酸酶抑制剂(CNIs)作为激素耐药型肾病综合征(SRNS)的初始治疗用药[8]。然而在本研究中,对出现TRPC6突变的3位患儿给予常见的钙调磷酸酶抑制剂药物(他克莫司和环孢素A),患儿的肾脏损伤症状未见缓解反而加重。现对3名患者的临床资料和基因测序结果进行如下报告,以探讨该现象发生的可能原因,并加强对该疾病的认知和理解。
病例资料
3例患儿均为汉族儿童,分别来自湖北省、陕西省、浙江省,患病均无家族史,其父母尿检均无异常,非近亲婚配。
病例1:男,于11个月龄时诊断为肾病综合征,初始给予每天2 mg/kg激素治疗4周,无明显好转,当地医院联合骁悉治疗,治疗3周依然无明显好转后改为联合1 mg/d的他克莫司治疗,后就诊我院。
病例2:女,于26个月龄时诊断为肾病综合征,发病初期行肾活检病理提示足细胞病变,肾小球免疫球蛋白(IgG、IgA)和补体(C1q、C3)的免疫荧光染色结果呈阴性,电镜结果提示FSGS,给予每天2 mg/kg的激素联合25 mg/d的环孢素A治疗后转至我院。
病例3:女,于9岁3个月诊断为肾病综合征,给予激素治疗4周,尿蛋白无明显好转,转至我院继续治疗。入院后行肾活检提示免疫荧光显示IgA、IgG、补体C3均为阳性,光镜检查提示肾小球局灶节段硬化,遂联合他克莫司治疗,剂量为1 mg/d。
一、用药及结果
病例1:按照1 mg/d的剂量给予他克莫司治疗,期间进行复查,结果显示患儿的Scr含量从35 μmol/L迅速升至119 μmol/L,且肾小管损伤加重;停用他克莫司,仅维持10 mg/d强的松治疗,结果发现患儿Scr浓度仍上升至307 μmol/L,同时出现少尿现象,遂给患儿以腹膜透析维持治疗。
病例2:给以每天2 mg/kg的环孢素A治疗,治疗期间患者的Scr迅速由25 μmol/L上升至174 μmol/L,遂给以血液透析维持治疗。
病例3:给予32 mg/d甲泼尼龙冲击治疗后联合1 mg/d他克莫司治疗,患儿24 h尿蛋白定量降至5 g/d后出院。随访期间患儿出现肌酐进行性升高的现象,由入院时的54 μmol/L升至420 μmol/L,后再次入院治疗,期间停药他克莫司,患者的肾功能损害一直未能缓解,在治疗4个月后进行腹膜透析维持。
二、基因检测结果
基因检测结果如表1。3位先证者的父母均表型正常且基因型与野生型相同,未出现变异。而3位患儿均发生突变,突变基因均为TRPC6基因,位于第11号常染色体上。
第1位患儿的突变位点位于第2 684位碱基,正常情况下该位点为鸟嘌呤(G),而先证者此处的碱基由鸟嘌呤(G)突变成胸腺嘧啶(T),对应的第895位氨基酸由精氨酸(Arg)突变为亮氨酸(Leu)。
第2位患儿的突变位点位于第523位碱基,正常情况下该位点为胞嘧啶(C),而先证者此处的碱基由胞嘧啶(C)突变成胸腺嘧啶(T),对应的第175位氨基酸由精氨酸(Arg)突变为色氨酸(Trp)。
第3位患儿的突变位点位于第329位碱基,正常情况下该位点为腺嘌呤(A),先证者的父母在此处的碱基与野生型相同,杂合概率为0;而先证者此处的碱基由腺嘌呤(A)突变为鸟嘌呤(G),对应的第110位氨基酸由天冬酰胺(Asn)突变为丝氨酸(Ser)。
3位先证者的临床表现均符合2型局灶阶段性肾小球硬化症的特征,该病遗传模式呈常染色体显性遗传。

表1 三位先证者基因检测结果
讨 论
FSGS的主要临床表现为蛋白尿,原因在于当患者的肾小球滤过功能受损时,血液中本来无法透过的大分子蛋白质进入尿液[9]。SD是肾小球滤过屏障的最外层,由相邻的足细胞足突交互形成,足细胞的结构和功能则由TRPC6与ACTN4、nephrin、podocin及CD2AP等多种SD蛋白共同维持[10-12]。当编码蛋白的基因发生突变时,将破坏SD的完整性进而打破肾小球滤过屏障,引起蛋白尿,最终发展为FSGS[6]。由此可见,TRPC6等基因的突变在FSGS的发生发展中起重要作用。
人TRPC6基因定位于第11号常染色体长臂 21区至 22 区,编码931个氨基酸[13-14]。当TRPC6基因发生突变时,多种SD相关蛋白间的相互作用将会被打破[11],进而引起足细胞的结构和功能受损,最终丧失其生物学功能。迄今为止,已证实TRPC6基因中的15处突变参与FSGS的发病[6](表2),其具体突变位点的位置如图1所示。可以看出,分别有1、2、1、1个突变发生于ANK的1、2、3、4区;6个突变位于胞质的C末端,2个突变位于胞质的N末端。
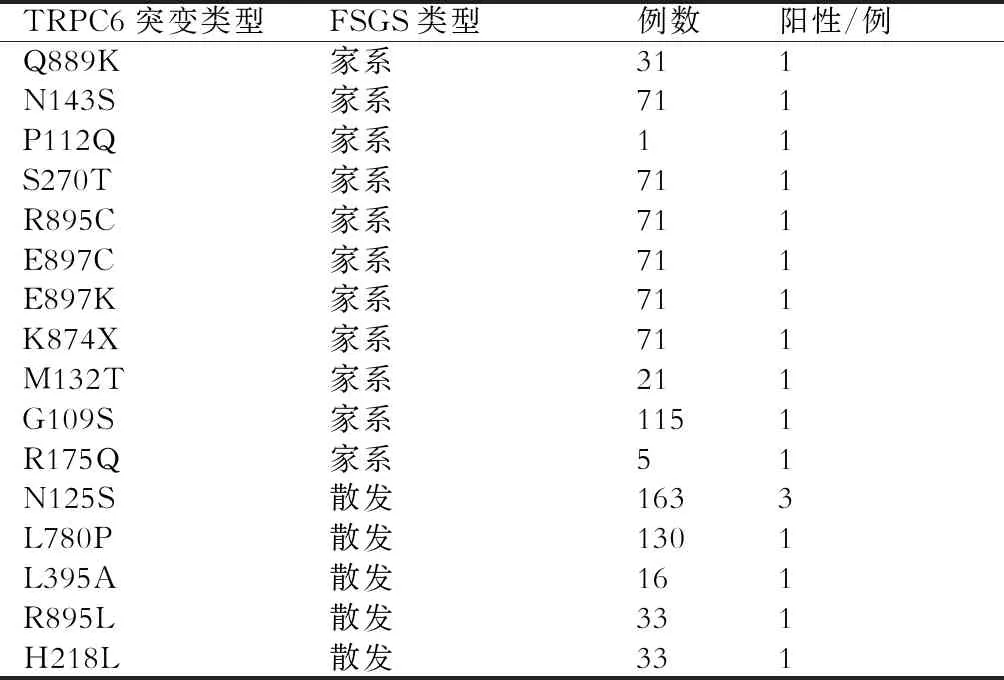
表2 TRPC6基因突变导致FSGS的研究总结
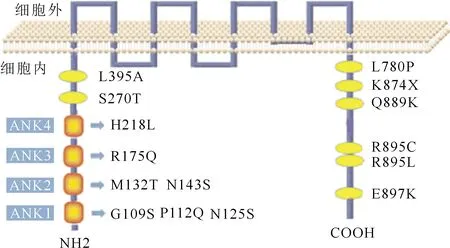
图1 TRPC6基因突变的位置
2005年Winn等[15]首次发现,当TRPC6发生P112Q突变时,可引起胞内的Ca2+浓度明显升高,最终引发家族性FSGS。截至目前,有包括R175Q等10个位点的突变均被证实可增加Ca2+的通道活性[16];随后,Reiser等[17]对71个家族性FSGS患者进行基因突变筛查,发现其中有5个单杂合突变,TRPC6基因突变率约为7%;另外,Kuang等[18]研究发现,TRPC6基因的第254位碱基由C突变成G与儿童激素抵抗性肾病综合征的发病密切相关;另有研究对80个FSGS中国家系进行筛查,结果发现有2个独立家系均出现Q889K突变,TRPC6的整体突变率为2.5%[19]。总之,TRPC6突变占中国家族性FSGS的比例约为3.2%,其他种族约为7%[16]。
本研究结果显示,3例患儿的父母均不发病且未携带致病基因,表明3例患儿的发病属于散发型FSGS。第1例患儿的突变为R895L,该位点已被Maddalena G证实[20];第2例患儿的突变为R175W,这与之前报道的家系型R175Q突变不一致[21];第3例患儿在9岁3个月发病,其第110位氨基酸由天冬酰胺突变为丝氨酸(N110S),该突变为国内外首次报道,其遗传方式也为常染色体显性遗传。由R175W和N110S突变引起FSGS的具体机制需要进一步地探讨。
Santin等[22]在非家族性的FSGS患者中发现,若患者出现N125S、G109S、L780P这3种突变,则使用钙调磷酸酶抑制剂能够有效的阻断钙调磷酸酶的活性,从而缓解蛋白尿的症状。另外,对于非TRPC6基因突变导致的肾病综合征,Mahmoud等[23]研究发现使用6.0 mg·kg-1·d-1的环孢素A治疗后,FSGS患者的完全缓解率超70%;Butani等[24]报道显示,16例SRNS患儿在接受他克莫司治疗4个月后,15例患者症状完全缓解。然而在本研究中,对激素耐药的3例患儿联合环孢素或他克莫司治疗后,患儿的Scr在治疗中和治疗后均迅速上涨,最后肾功能非但未缓解反而损伤加剧。针对此现象,我们推测,这可能是由于患儿的突变基因影响了钙调磷酸酶抑制剂的吸收和代谢药动学[19],进而导致Scr浓度大幅上升,但具体作用机制还有待进一步验证。
综上所述,本研究提供了两个新的FSGS突变位点,即R175W和N110S。两例患儿基因突变的原因及其与FSGS的确切关系需要更多的体外功能试验甚至动物模型试验来明确。另外,在治疗FSGS患者时,需要综合考虑发病原因、药物疗效、自身实际情况等多方面因素,选择合适的免疫抑制剂类药物和剂量,以达到安全有效的治疗目的。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
中国药学药品知识仓库(2022年5期)2022-04-11
皮肤病与性病(2021年3期)2021-07-30
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
西南军医(2016年5期)2016-01-23
