
熟石膏陈化过程的研究进展
2020-09-10 10:08刘元会唐晓娜姜小鹏张云波
无机盐工业 2020年9期
刘元会,唐晓娜,谢 蕾,姜小鹏,张云波
(青岛量子元基环保科技有限公司,山东青岛266000)
陈化泛指体系经历一个过程后放置一段时间进行其他过程, 尤其指在固体沉淀过程中待沉淀完全后使溶液在一定条件下静止存放一段时间, 目的是令里面的组分得到充分反应或令悬浮物沉降。 许多陶瓷制品[1-2]和水泥[3]等材料也需要在空气中水蒸气的作用下陈化以改善其性能。
对于一般刚煅烧出来的熟石膏, 其物相组成不稳定、内含能量较高、分散度大、吸附活性高,从而出现调制后标稠需水量大、 强度低及凝结时间不稳定等现象。 改善这种状况的办法是使煅烧得到的熟石膏在密闭的料仓中存放,利用物料的温度(380 K 以上)可以使物料中残存的二水石膏(CaSO4·2H2O)吸热进一步转变为半水石膏(β-CaSO4·0.5H2O),同时其中的可溶性无水石膏(AⅢ-CaSO4)也可以吸取物料周围的水分转变为半水石膏(β-CaSO4·0.5H2O),这种在一定温度和湿度下相组分的转变以及晶体的某些变化就是熟石膏陈化的实质。 熟石膏通过陈化均化,既可以稳定粉料物相的组成和性能,又可以提高有益物相组成的比例, 从而提高和改善粉料的工艺和物理性能,提高产品质量。
1 熟石膏陈化基本原理
熟石膏陈化一般分为自然陈化和强制陈化。 强制式石膏陈化均化装置不仅具有占地面积小、 陈化均化能耗低、成本低的优点,还能够提高陈化均化的效率和效果。 强制式石膏陈化均化装置在国内外经历了一段历史的变革,版本不断更新[4-5]。
石膏在陈化过程中物理性质的变化已有作者进行了研究。 李逢仁[6]比较系统地总结了各种熟石膏在不同陈化条件下物理性质的变化规律, 详细分析了熟石膏陈化过程中发生相变的基本类型及影响因素。作者提到在陈化初期有再生二水石膏的生成,再生二水石膏的生成与脱水对熟石膏的强度、 凝结时间的变化产生显著影响。 作者探讨了熟石膏陈化过程中发生的相变与水化性质及物理性质变化之间的关系。 马咸尧等[7]研究了二水石膏脱水后在363~373 K 保温均化和在室温存放陈化时间效应对β 半水石膏性能的影响。 经回转窑脱水的石膏粉须进行均化处理以达到相组成稳定、材料性能稳定。作者发现均化时间对β 半水石膏强度的影响有一最佳值,均化时间一般以30 h 为宜, 这时材料的强度最高、孔隙率最小。
余红发等[8]在293 K 和相对湿度为50%的室内条件下进行了石膏脱水相陈化动力学机理研究,研究陈化过程中相组成的变化规律, 建立了可溶性CaSO4的陈化动力学方程, 用该方程式能够确定石膏脱水相进入陈化稳定期的时间。 其动力学机理包括水化反应和扩散过程,其陈化过程在初中期(α<70%)受化学反应控制,中后期(α=70%~90%)受杨德尔扩散控制,晚期(α>90%)受金斯特林格扩散控制。 在陈化过程中,石膏脱水相的物理力学性能要经历不稳期、稳定期和失稳期,当可溶性CaSO4完全转化成β-CaSO4·0.5H2O 时进入陈化稳定期,此时用水量少、凝结快、强度高。
2 熟石膏陈化过程中水蒸气在AⅢ-CaSO4上的物理吸附与化学吸附
2.1 水蒸气在AⅢ-CaSO4 上的物理吸附
Preturlan 等[9-10]测量了不同温度和分压时水蒸气在AⅢ-CaSO4上的吸附数据(见图1),讨论了吸附层特征与吸附现象的热力学本质。 他们基于吸附等温线图提出了局部化单层吸附模型,关于建模过程吸附层被认为是自由位点与吸附位点之间的表面溶液。 最初,他们假设表面溶液的理想行为并采用Langmuir 吸附模型拟合,但是这种模型不足以表示吸附等温线的完整性,并且拟合参数出现了一些分散。 随后他们提出了表面溶液非理想行为的假设,当与理想行为的差异不明显时考虑了正则溶液假设。因此引入了一个参数来考虑在表面溶液组分之间的交互作用,该程序在描述实验数据时更准确,见式(1)。 对于这两种方法,吸附能量的值相当低,并证实了初始物理吸附的假设。 关于水蒸气在AⅢ-CaSO4上的物理吸附文献报道内容较少,然而水蒸气在其他固体上的物理吸附有深入广泛的研究[11]。

图1 CaSO4 总水含量等压线
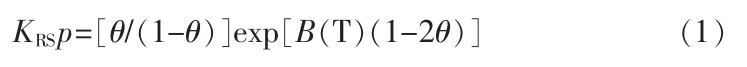
式中:KRS是规则溶液吸附平衡常数;p 是压力;θ 是覆盖范围;B(T)是依靠温度和溶液中分子间相互作用的参数。
2.2 水蒸气在AⅢ-CaSO4 上的化学吸附
Tang 等[12]用Monte Carlo(MC)方法模拟了水蒸气在可溶性无水石膏(AⅢ-CaSO4)晶格隧道内的吸附行为, 计算的水化学吸附等温线结果和实验值一致性很好,也显示了分布在AⅢ-CaSO4框架隧道内水分子的构造。 研究显示AⅢ-CaSO4在相当低的相对湿度和温度范围(215-298 K)内有极好的吸附水蒸气的能力。当相对湿度接近1%、温度为298 K 时,AⅢ-CaSO4能够吸附空气中的水蒸气再水合形成CaSO4·0.5H2O。 然而当温度为298 K、相对湿度超过80%时,AⅢ-CaSO4晶格内结合的水分子数达到0.67。 当水蒸气分压从0 Pa 增至0.04 Pa、 温度为215 K 时,结晶水数值n 从0 增至0.5。
AⅢ-CaSO4晶体陈化过程中吸收空气中的水蒸气分子化合成β-CaSO4·0.5H2O 晶体而放出热量,AⅢ-CaSO4陈化放热这一现象有重要的应用。 例如Lee 等[13]提 出 采 用 可 逆 硫 酸 钙/水(CaSO4/CaSO4·1/2H2O)反应制备化学热泵(CHP)材料,以有效利用废能源, 该系统是有效利用能源的重要技术。 依据CHP 体系,Lee 等提出了一种新型的卡车制冷系统,该系统利用CHP 将发动机余热储存起来,并将其作为冷热循环/释放出来,以供制冷室在怠速停车即发动机停止时冷却/制冷。 然而在CHP 制冷模式的实验中, 在500 次左右的重复实验中,CHP 材料有时会出现水化/脱水反应的退化现象,Lee 等解释了退化的主要原因和解决方法。
AⅢ-CaSO4晶体吸收空气中水蒸气分子的反应属于气固相反应[14],水蒸气分子在AⅢ-CaSO4晶体纳米通道内物理传输[15]并发生化学吸附形成β-CaSO4·0.5H2O 晶体。 然而没有文献报道水蒸气分子在AⅢ-CaSO4晶体纳米通道内发生化学吸附反应的具体过程, 只有其他的一些化学吸附现象被学者们研究。 例如周斌[16]利用第一性原理研究了水分子化学吸附对碳纳米管电学性质的影响以及水分子在碳纳米管表面的化学吸附过程。 王娟[17]以高岭石吸附量较大的Pb(Ⅱ)、Cu(Ⅱ)、Zn(Ⅱ)为例,从微观角度确定其在水体环境中的溶剂化结构, 并在此基础上对水合重金属离子在高岭石(001)晶面的化学吸附进行研究。 Kumar 等[18]研究了半导体金属氧化物表面有湿度存在下氧气和还原气体的化学吸附的数字模型,在模型中用Wolkenstein 吸附理论代替传统的Langmuir 等温线进行氧气、还原气体(CO)分子和水蒸气化学吸附的数值模拟。
3 熟石膏陈化过程中CaSO4·2H2O 晶体的脱水行为与脱水动力学
3.1 CaSO4·2H2O 晶体的脱水行为
Chang 等[19]用热拉曼光谱从298~573 K 在静止空气条件下进行了CaSO4·2H2O 和CaSO4·0.5H2O 脱水过程的原位监控研究。 热拉曼光谱表明了样品结构 的 变 化,波 段 强 度 说 明 了CaSO4·2H2O、CaSO4·0.5H2O、CaSO4含量的变化。 这与从热重分析(TGA)获得的温度记录图非常相似, 其优点是每种物种的数量都可以单独测量。 能带强度的导数给出了转换中的最大速率。 在热拉曼光谱图中观察到了新形成物种和旧物种数量的变化, 而不是差热分析中的一个峰。波段位置的变化也显示了这些变化。样品的热拉曼光谱图表明,在实验条件下CaSO4·2H2O 的脱水是两步连续反应,因为3 个物种在小的温度范围连续出现,CaSO4·0.5H2O 是中间产物。Putnis 等[20]在可忽略的水蒸气分压下用原位红外光谱(IR)和热重分析研究了石膏(CaSO4·2H2O)的脱水行为,来测定是否有中间相(CaSO4·nH2O)存在。 热重分析显示了连续的水减少具有的活化能为90.3 kJ/mol, 活化能作为脱水度的函数没有发生变化,脱水行为是一种相界控制反应。 IR 图明确显示了3 个分离相CaSO4·2H2O、CaSO4·0.5H2O、γ-CaSO4的存在,并且随着脱水的进行每一个连续相晶核的形成。在IR 图中,对于CaSO4·2H2O→CaSO4·0.5H2O 结构的变化,SO4四面体的弯曲振动相比于OH 伸缩区更加敏感;对于CaSO4·2H2O→CaSO4·0.5H2O 结构的变化,在1 622 cm-1处的弯曲振动没有改变位置,当CaSO4·0.5H2O 脱水时没有发现OH 伸缩振动的分裂。 他们没有发现任何中间水含量相的存在。 Abriel 等[21]用中子和X 射线粉末衍射在295~623 K 研究了石膏的脱水行为,用背景强度作为水含量的测量值。 Bragg 峰产生了4 个主要的相CaSO4·2H2O、CaSO4·(H2O)x(0<x<1)、AⅢ-CaSO4、AⅡ-CaSO4, 在高的水蒸气分压测量出了最大水含量x=0.74 的子水合物。 半水合物的转调点显示了进一步的脱水过程要求更高的能量破坏CaSO4结构框架的氢键。 在隧道结构内的水化和脱水机理是强烈的拓扑, 反应中主结构的三维网络被保护。McAdie[22]研究了在水蒸气分压下CaSO4·2H2O晶体的脱水行为, 脱水从晶体表面的晶核形成开始而不是从位错开始, 脱水过程一直持续到分离的反应区域交叠, 这一过程通过个别晶体和脱落晶面扩展。 大量脱水表现为线性、 零级关联, 这显示了成核-扩展机理发生在一个相当恒定的反应表面,在此获取其他潜在核的影响不再重要。CaSO4·2H2O 的脱水速率随着水蒸气分压的提高有规律地降低。
3.2 CaSO4·2H2O 晶体脱水动力学
Ballirano 等[23]利 用 实 时 实 验 室 平 行 束X 射 线粉末衍射数据进行了空气中石膏脱水的热行为和动力学研究,并且这些数据由Rietveld 方法评价。分析了298~373 K 的热膨胀, 用Einstein 方程计算出晶胞边和晶胞体积的高温极限分别为4.29×10-6、4.94×10-5、2.97×10-5、8.21×10-5。 石膏的热膨胀具有很强的各向异性,沿b 轴方向的热膨胀较大,主要是由于H2…O1 氢键的弱化所致。 石膏脱水在等温条件(348~403 K)升温5 K 进行,脱水过程经历CaSO4·2H2O→CaSO4·0.5H2O→γ-CaSO4步骤,用Avrami方程(2)拟合实验数据计算了该过程的经验活化能(Ea)为109(12)kJ/mol,同时得到了不同温度的速率常数,在所分析的温度范围内没有观察到相变机制的变化。 Cave 等[24]进行了流化床反应器中石膏脱水动力学研究,在流化床反应器中在床温为373~443 K 条件下测定了粒径为35~67 μm 的脱硫石膏的脱水速率。流化气体为空气,由0.1~35.5 kPa 的水蒸气和二氧化碳组成。结果表明,所有这些条件下的脱水动力学都可以用二维Avrami-Erofeev 表达式来模拟。在413 K 煅烧温度下,无论脱硫石膏颗粒的粒径大小, 反应速率与水蒸气压力之间的唯一关联是明显的。在此条件下,还证明了转化率与简化反应时间之间的唯一关系。 Lou 等[25]在非等温和等温条件下研究了脱硫石膏在N2气氛(自生和可忽略水分压)条件下的脱水行为及动力学。石膏脱水经过一步(CaSO4·2H2O→γ-CaSO4)或两步(CaSO4·2H2O→CaSO4·0.5H2O→γ-CaSO4)取决于温度和水分压。一步过程发生在非等温条件可忽略水分压以及等温条件373 K 以下, 然而两步过程发生在非等温条件自生水分压和等温条件373 K 以上。 3 种脱硫石膏在脱水行为上的差异很可能是由于不同的结晶特性(粒度和习性)和杂质。 用两种“模型自由”动力学方法对非等温分析的实验数据进行了分析,用Avrami和线性方程进行了等温分析。 表观经验活化能(Ea)表明, 石膏向半水合物的转变主要受成核和生长机制控制,而石膏向γ-硬石膏的转变则以相界机制为主。Carbone 等[26]通过能量分散X 射线衍射(EDXD)在减压(100 Pa)及313~353 K 研究了石膏(CaSO4·2H2O)的脱水动力学。 这个脱水过程遵循JMAK(Johnson-Mehl-Avrami Kinetic)模型。 Arrhenius 表达式的拟合过程提供了75.35(8.37)kJ/mol 活化能。在这些实验条件下,脱水过程通过一步转换路径进行,即CaSO4·2H2O→γ-CaSO4。CaSO4·0.5H2O 在相同条件的脱水实验显示了一个较快的过程,即γ-CaSO4是最终产物。根据CaSO4·0.5H2O 和γ-CaSO4之间的结构关系, 脱水过程应该通过水分子沿c 轴隧道的逸出发生,服从一维行为。因此暗示了成核和新相成长的Avrami 模型不能被应用。Ball 等[27]在353~425 K、水蒸气分压为1.333×10-6~6.0 kPa 条件下研究了CaSO4·2H2O 的脱水动力学。反应产物受温度和分压的控制, 并且计算了一些过程的活化能。 温度大于383 K 时, 整个分压范围内脱水过程受扩散机制控制, 在实验和计算简化时间曲线之间最好的拟合是依据一维扩散并且遵从抛物线法则。但是,在较低温度(<383 K)时动力学行为复杂,很容易被水蒸气影响,产物成核和边界控制很重要。 在较低温度、水蒸气分压>0.133 3 kPa 时, 成核是控速步骤, 遵循Avrami-Erofeev 方程(3)。 随着温度升高,边界控制过程成为主要方式,其动力学遵从‘contracting-disc’方程(4)。 在较低分压(1.333×10-6~0.106 6 kPa)时成核控制没有观测到,反应遵从‘contracting-disc’方程直到扩散成为控速步骤。
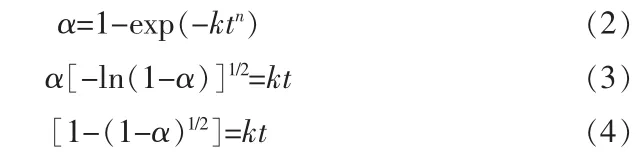
式中:α 是反应转化分数;k 是速率常数;t 是反应时间;n 是反应指数。
4 熟石膏陈化过程中CaSO4·2H2O、CaSO4·0.5H2O、AⅢ-CaSO4 晶体结构
4.1 CaSO4·2H2O、CaSO4·0.5H2O、AⅢ-CaSO4 晶体结构
CaSO4·2H2O 空间群为, 单斜晶系,其结构可视为具有配位数8 的钙离子联结硫酸根四面体构成双层结构层,H2O 分子分布在双层结构之间。离子结合层内部是由正负离子相互作用而产生的结合力; 水分子层内部是由偶极子与偶极子相互作用而产生的结合力; 离子结合层与水分子层之间则是由离子与偶极子的相互作用而产生的结合力。 Ca2+与相邻的4 个中的6 个O 原子和2 个水分子的O 原子相连接。 结构层与(010)面网平行,因此石膏典型的晶体形态为板状{010}。 由于双层结构是通过水分子微弱的氢键联接,二水石膏在(010)晶面上具有完全的解理[28]。
半水石膏CaSO4·0.5H2O 具有高伪对称结构以及不能获得精确的衍射数据, 因而出现不同的半水石膏的晶体学参数。总体来说,α 型半水石膏的晶体结构存在的争议主要集中在结晶水的数量及其占据的空间位置,空间群是P3121、I121 还是C121。 这是由于α 型半水石膏单晶体制备方法的不同,造成了结晶水含量及其在晶格中占据的位置发生了变化[28]。
AⅢ-CaSO4晶格由Ca2+和四面体构成,在链条-Ca-SO4-Ca-SO4-里交替的Ca2+和沿[001]方向形成直的连续链条,结果这些链条呈现周期排列,显示了蜂窝状结构。 这蜂窝状空穴赋予AⅢ-CaSO4吸附空气中水分子的能力,形成CaSO4·nH2O 相。 AⅢ-CaSO4和CaSO4·0.5H2O 的 结 构 高度相似,它们的晶格都提供了蜂窝状的隧道,具有0.3 nm 直径[29]。
Nagore 等[30]测量了CaSO4·2H2O、CaSO4·0.5H2O和AⅢ-CaSO4晶体的主要Raman 光谱带分别是1 008、1 015、1 025 cm-1。 3 种 晶 体 结 构 示 意 图 见图2。 从图2 可见,陈化过程中石膏CaSO4·2H2O 脱水变为半水石膏CaSO4·0.5H2O 时,石膏晶体结构发生了较大变化; 而AⅢ-CaSO4吸收水蒸气生成CaSO4·0.5H2O 时,二者的结构几乎相同。
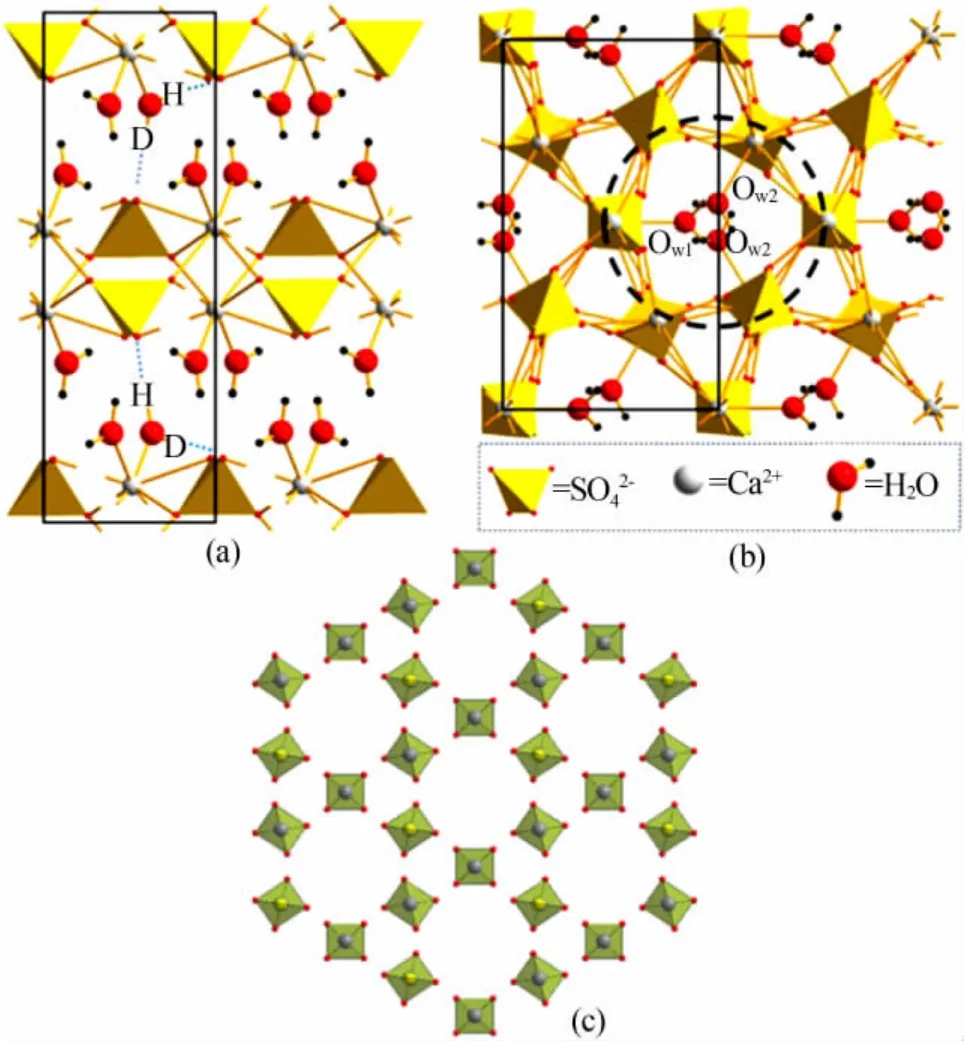
图2 CaSO4·2H2O(a)、CaSO4·0.5H2O(b)、AⅢ-CaSO4(c)晶体结构示意图
4.2 CaSO4·2H2O 和CaSO4·0.5H2O 晶体中水分子结构信息
Yan 等[31]研究了CaSO4·2H2O 和CaSO4·0.5H2O晶体中水分子运动的信息。 两种水合物晶体中水分子个数的不同对水分子动力学有显著影响。 二维红外光谱(2D IR)测量显示了新制备的CaSO4·2H2O 中水分子排列存在少量的不均匀性, 水分子氢氧基伸展模式在晶体退火之前有很小的非均匀扩展程度,在348 K 退火后水分子变得高度有序。 极化选择泵探针光谱(PSPP)仅仅观测到了CaSO4·2H2O 晶体中惯性定位松弛, 水分子不经历任何扩散取向弛豫。CaSO4·2H2O 晶体中的水分子被约束为晶格方面的单一构象,水分子运动发生在极短的时间范围内,这很可能是因为整个晶格经历了在室温由热填充的声子引起的结构波动。相反在CaSO4·0.5H2O 晶体管状隧道中的水分子动力学上混乱。如2D IR 数据所示,水羟基拉伸吸收光谱除了具有均相组分外, 还具有明显的非均匀展宽。 非均匀展宽显示水分子住在许多稍微不同的晶格构造中, 尤其是在通过氢键连接羟基的硫酸盐构造中。 2D IR 实验显示在298 K 有一个非均匀展宽组分,它经历了十几个ps 的光谱扩散。光谱扩散由结构演变引起,在这个演变中水分子汲取了许多构象。
Mandal 等[32]用IR 数 据 和XRD 数 据 解 释 了CaSO4·2H2O 和CaSO4·0.5H2O 晶体中松散容纳和强烈容纳的两类水分子结合的本质。CaSO4·2H2O 晶体的红外吸收光谱中水分子在1 680、1 620 cm-1处的两个弯曲振动峰是由于存在两类水分子,1 680 cm-1处归属于松散容纳水分子,1 620 cm-1处由强烈容纳水分子(阴离子水)引起。 CaSO4·0.5H2O 晶体仅包含强烈容纳的阴离子水,拥有唯一的一个弯曲振动1 620 cm-1。 两个晶体中1 620 cm-1处阴离子水通过氢键附属到阴离子上。 CaSO4、CaSO4·0.5H2O、CaSO4·2H2O 晶体的红外吸收光谱中依次拥有弯曲振动(675、610、595 cm-1)(650、580 cm-1)(670、600 cm-1)。 弯曲振动吸收模式的变化显示了构象的变化, 这归因于通过邻近水分子的扰动。因此CaSO4·2H2O 晶体中水分子和吸收带的红外光谱研究显示了在CaSO4·2H2O 晶体中有两类不对称的水分子,3/2 水分子(松散容纳)附属到Ca 原子,1/2 水分子(强烈容纳)通过氢键容纳到SO42-。
5 结论与展望
通过上述分析发现熟石膏陈化过程中水蒸气在AⅢ-CaSO4纳米通道内的化学吸附反应过程以及CaSO4·2H2O 晶体的脱水过程,文献没有从微观角度进行反应历程的具体研究。 因此我们还有很多工作要做,继续运用实验工具和分子模拟软件研究AⅢ-CaSO4纳米通道吸收水蒸气和CaSO4·2H2O 晶体脱水的微观反应机理。 例如水蒸气与AⅢ-CaSO4晶体颗粒的微观反应, 实验研究运用梯度脉冲核磁共振技术(PFG-NMR)测量客体H2O 分子在AⅢ-CaSO4纳米通道内的受限运动扩散系数, 应用吸附速率法测定H2O 分子在AⅢ-CaSO4纳米孔晶体的表面势垒和渗透率。 可以研究水蒸气中H2O 分子进入AⅢ-CaSO4晶体纳米通道所发生的物理扩散传质输运行为, 但是一个水分子与Ca2+和化学结合后是否会阻塞纳米通道内其他水分子通过, 具体的化学吸附反应过程很难用实验仪器观察,可以借助分子模拟软件NAMD 软件或LAMMPS 软件来实现。
石膏的脱水分解反应式看起来相当简单, 但是其中所包含的微观动力学机制相当复杂。 可以用高温显微镜从亚观水平直接观察二水石膏的脱水过程,例如在二水石膏晶片最薄弱部位如缺陷、空洞、解理和裂纹等处观察水分子在这些部位的迁移和水的出露点的形成以及围绕核点聚集的半水石膏微晶体的长大过程。 进一步从微观水平用高温原位拉曼光谱测量CaSO4·2H2O 脱水分解过程中各种振动峰的强度变化和振动频率位移情况,用中子衍射追踪石膏分解过程的结构演变及其衍射图像的变化,更加具体形象微观的分解化学行为和物理行为可以借助分子模拟软件NAMD 软件或LAMMPS 软件来实现。
猜你喜欢
化工学报(2022年10期)2022-11-13
科普童话·学霸日记(2021年4期)2021-09-05
农家参谋(2021年5期)2021-06-20
科教新报(2021年11期)2021-05-12
意林原创版(2017年11期)2017-12-01
红蜻蜓·低年级(2017年10期)2017-11-21
