
微反应器研究陈化过程对铜锰催化剂的影响
2022-11-13 07:31方凯伦陈帅帅付家崴蒋新
化工学报 2022年10期
方凯伦,陈帅帅,付家崴,蒋新
(浙江大学化学工程与生物工程学院,浙江 省化工高效制造技术重点实验室,浙江 杭州 310027)
引 言
Cu-Mn 催化剂在VOCs 催化氧化方面的良好性能使其在大气污染物治理方面具有广阔的应用前景[1-2]。目前制备Cu-Mn 复合氧化物催化剂最常用的方法是共沉淀法[3],通常是在半分批式的搅拌釜反应器中完成,具体流程为:金属盐与碱性溶液混合发生沉淀反应得到沉淀物,沉淀物在母液中经过一段时间的陈化,再经洗涤、过滤、干燥、焙烧得到催化剂。
在陈化过程中,无定形的沉淀物会通过结构重排形成晶态产物,这一变化过程对后续的催化剂结构形成会产生较大的影响[4-5]。虽然大部分研究者在制备Cu-Mn 催化剂的流程中都采用了陈化过程,然而针对陈化过程本身的研究却很少。以往对于陈化过程的研究显示[6-7],随着陈化时间的延长,Cu-Mn催化剂性能总体上先变差后变好,长时间陈化后制得的催化剂性能优于未陈化的催化剂。然而,这些研究并没有揭示陈化过程的机制,对沉淀物在陈化过程中的结构变化过程缺乏深入的认识。同时,文献中传统共沉淀法均在搅拌釜中进行反应,采用半间歇操作,形成的沉淀物已经不可避免地发生了一段时间的陈化,因此,无法对陈化最初的阶段进行研究。
针对这些问题,本文采用连续式的微反应器通过共沉淀法制备Cu-Mn 催化剂。利用微反应器的快速混合及微通道高效热交换的特点[8-9],在极短时间内完成反应过程并迅速冷却,以制备未陈化的沉淀产物;同时,短时间的陈化过程直接在延长管路中完成,可以得到陈化时间在分钟级的产物,以研究陈化过程中沉淀物结构演变过程及其对后续催化剂的影响。
1 实验材料和方法
1.1 样品的制备
制备Cu-Mn催化剂的共沉淀过程在图1所示的反应装置中进行,图中示出Caterpillar 微反应器的结构,微通道中亚毫米级的扰流结构极大增强了溶液的混合效果。样品的具体制备流程如下。反应溶液在预热盘管中加热至70℃后进入Caterpillar 微反应器,本文实验条件下反应液在微反应器通道中停留约10 ms。流出Caterpillar 微反应器后,物料进入陈化段,不同陈化时间采用不同设计:对于不陈化的实验,反应液流出微反应器后直接进入置于冰水浴的盘管,迅速降温避免陈化,得到未陈化的沉淀物;对于陈化30 s 及1 min 的实验,反应液从微反应器流出后先进入80℃水浴中的延长段管路,通过管路长度控制陈化时间,再进入置于冰水浴的盘管,快速降温终止陈化过程;对于陈化2 min 及以上的实验,首先通过反应器出口80℃水浴下的延长段管路陈化1 min 后,再进入80℃水浴下的烧瓶,完成剩余陈化后迅速冷却。随后将沉淀物洗涤、过滤后放入90℃烘箱烘干24 h,沉淀物样品按照陈化时间进行命名,如P-0 min、P-1 min、P-360 min。沉淀物在410℃下焙烧2 h 得到氧化物催化剂样品,样品按照陈化时间进行命名,如C-0 min、C-1 min、C-360 min。另用上述相同方法制备2 组纯组分Mn 沉淀物,一组不陈化,另一组陈化360 min,作为对照组样品进行表征分析。

图1 样品制备流程图Fig.1 Flow diagram of preparation process
实验中采用平流泵控制混盐溶液的流量为40 ml·min-1,通过调整碳酸钠溶液的流量将出口pH 保持在8.0±0.2。Cu(NO3)2和Mn(NO3)2的混盐溶液总浓度为0.2 mol·L-1,Cu∕Mn 摩尔比为1∶4,Na2CO3溶液浓度为0.2 mol·L-1。实验中的管路均以盘管的形式置于恒温水浴锅中,以实现对温度的精确控制。
1.2 样品的表征
X 射线多晶衍射(XRD)仪器型号为X'Pert PRO MPD,光源为Cu Kα射线,对应波长0.15406 nm。扫描范围和速率分别设定为10°~80°和5(°)·min-1。
高倍透射电镜(HRTEM)仪器型号为JEOL JEM 2100F,采用无水乙醇作为分散剂将待测样品在超声下处理15 min,将处理后的分散液负载于钼网超薄微栅膜上,干燥后的样品通过高倍透射电子显微镜进行形貌和元素分布的分析。
拉曼光谱仪器型号为雷尼绍Renishaw in Via,取50 mg 样品粉末进行结构表征,激光器波长532 nm,测试波数范围为50~3000 cm-1。
热重分析(TGA)仪器型号为TGA∕DSC3+(瑞士梅特勒同步热分析仪),在氧化铝坩埚中称量8~10 mg 粉末样品,在10℃·min-1的升温速率下对其热分解特性进行分析,测定的温度为50~700℃,选择空气作为测试气氛。
X 射线光电子能谱分析(XPS)仪器型号为Thermo Scientific K-Alpha,称取20~30 mg 样品粉末进行测试,光源为Al Kα射线(hv=1486.6 eV),电极的电压和电流分别为12 kV和6 mA。
1.3 催化活性考评
采用甲苯催化降解反应来考评Cu-Mn 催化剂的催化活性。反应装置为固定床反应器,加热炉中的高硼硅玻璃管内径为4 mm,长180 mm。催化剂的装填分为三部分,下层为1 g 石英砂作为支撑,中层为0.1 g催化剂与0.5 g石英砂的均匀混合物,上层为0.5 g 石英砂。实验中一路空气鼓泡通过冰水浴中的甲苯,与另一路稀释空气混合,通过调整两股气体的比例使甲苯浓度为0.1%,控制进入固定床反应器的总体积流率为30 ml·min-1,空速GHSV=18000 ml·g-1·h-1,常压反应。
催化活性以甲苯的转化率作为指标,含量分析采用气相色谱,型号为福立公司GC9790Ⅱ。色谱柱为RB-5 型毛细色谱柱(柱长30 m,内径320 μm,膜厚0.5 μm),柱温80℃,以氮气为载气,流速25 ml·min-1。色谱柱出口采用FID 检测器检测甲苯含量,检测器温度150℃。
2 实验结果与讨论
2.1 陈化过程对催化剂性能的影响
微反应器制备得到的Cu-Mn 共沉淀物,陈化后焙烧得到催化剂。通过甲苯催化降解反应考察陈化时间对催化剂活性的影响,结果如图2 所示。可以看到,未陈化沉淀物制得的催化剂C-0 min 的催化活性明显优于其他催化剂,T90=193℃(T90为甲苯达到90%转化率时的温度)。随着陈化时间的延长,催化剂活性快速下降,在陈化时间为5 min 时,催化剂活性最差。随后,陈化时间再延长,催化活性小幅上升,最终T90稳定在210℃左右。

图2 不同陈化时间的Cu-Mn催化剂甲苯催化性能Fig.2 Toluene catalytic performance of Cu-Mn catalyst with different aging time
陈化过程对Cu-Mn 催化剂的影响,文献中研究较少。在Mirzaei 等[6]和Hutchings 等[7]的研究中也发现Cu-Mn 催化剂性能随着陈化时间的延长出现先变差后变好的现象,这与本文的研究结果一致。然而,在他们的研究中,未陈化的催化剂活性不如长时间陈化的催化剂的活性,这与本文结论相反。仔细分析这些研究工作可以发现,他们的催化剂制备是在搅拌釜反应器中进行的,这导致先期形成的沉淀物经历了一段时间的陈化过程,因此,这些文献中所认为的未陈化样品,实际上已经经历了一段短时间的陈化。由图2 可以看到,这段短时间陈化过程会导致催化剂活性迅速下降,使得研究者认为未陈化样品活性不好。可能正是由于类似的原因,大部分研究者都把重点放在了经历较长时间陈化的催化剂上[6-7,10],忽略了未陈化催化剂的研究。为探究未陈化催化剂性能优异的原因及催化剂活性随着陈化过程在短时间内迅速变化的原因,对不同陈化时间的沉淀物和催化剂样品的物相及结构进行了表征和分析。
2.2 陈化过程对沉淀物的影响
通过XRD 分析沉淀物物相,结果如图3 所示。未陈化的Cu-Mn 沉淀物结晶度很差,仅在31°和52°附近出现微弱的衍射峰,陈化后的Cu-Mn 沉淀物均在24°、31°和52°附近出现了稳定特征峰,与陈化后的纯Mn 沉淀物相同,分别对应于MnCO3的(012)、(104)和(116)晶面[11]。相对于P-0 min 样品,可以看到短时间的陈化就使样品的结晶度迅速变高,这表明Cu-Mn 沉淀物的陈化过程非常迅速。进一步观察可以发现,P-0 min 样品的XRD 谱图中,31°左右的衍射峰已开始显现,表明该样品已经经历了少量的陈化。这一现象说明即使是微反应器极短的反应时间也难以保证产物完全不陈化,而文献中的搅拌釜反应过程就更难避免陈化的发生了。
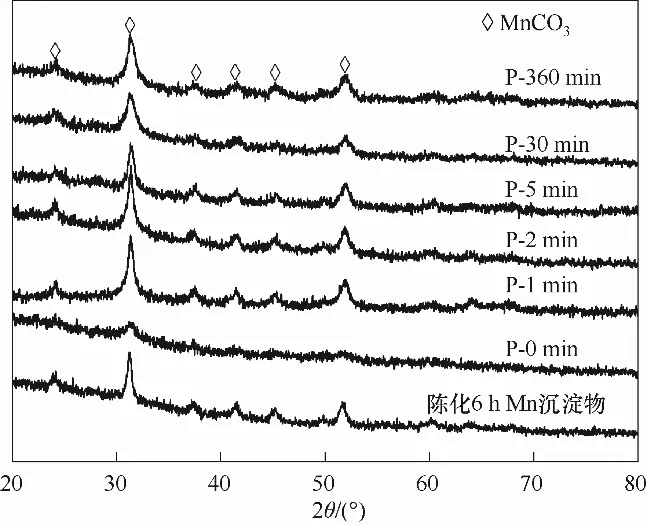
图3 不同陈化时间沉淀物的XRD谱图Fig.3 XRD patterns of precipitates at different aging time
不同陈化时间样品的XRD 谱图中均未出现与Cu 元素有关的物相衍射峰,这可能是由于Cu 晶相的形成速率较慢以及Cu 含量偏低,且部分Cu2+进入了MnCO3物相[11-12]。根据XRD 衍射峰的半高宽可以得到MnCO3晶粒的平均粒径,P-1 min~P-360 min样品的平均晶粒粒径分别为12.6、11.1、13.0、9.0、9.0 nm。这表明随着陈化时间的延长,晶粒粒径出现了反常的下降趋势,这可能与Cu2+进入了MnCO3物相有关。
对上述样品进行拉曼分析(图4),发现随着陈化的进行,对应于CO2-3的对称伸缩振动v1(CO2-3)[13-14]在1086 cm-1附近的特征峰先变强,至5 min 左右时达到最大,随后开始变弱;对应于CO2-3的面外弯曲振动[15]在291 cm-1附近的特征峰也呈现出类似变化过程;而归属不明的在614 cm-1附近的特征峰则出现相反的变化规律,即首先变弱,5 min 左右时几乎完全消失,随后又开始变强。从拉曼谱图中这几个峰的变化过程可以看出,在陈化过程中样品结构的变化并不是一个线性的演变过程,表明了陈化过程的复杂性。

图4 不同陈化时间沉淀物的拉曼谱图Fig.4 Raman spectra of precipitates at different aging time
2.3 陈化过程对沉淀物热分解过程的影响
沉淀物的热重分析结果如图5 和图6 所示。陈化后样品的分解主要集中在400~500℃,而未陈化的P-0 min 样品DTG 曲线在300℃左右就出现明显的失重峰,同时P-0 min 样品的TG-MS 表明在此区域有CO2和H2O 逸出,但两者并不重合。由于极快速沉淀反应形成的沉淀物,难以在未陈化条件下形成有序结构,因此P-0 min 样品主要由无定形物质构成。以往研究表明,Cu2+单独沉淀的产物主要是碱式碳酸铜,其分解温度在300℃附近[12],因此P-0 min 样品在300℃左右可能存在碱式碳酸铜的分解。根据投料Cu 与Mn 的摩尔比为1∶4,计算碱式碳酸铜分解的失重率约为3%,而P-0 min 样品在100~300℃的失重率达到了12.84%。考虑到未陈化的碳酸锰分解温度也在300℃左右[图6(a)],认为300℃左右也存在着碳酸锰的分解。此外P-0 min 样品的DTG 曲线中400~500℃范围内还有一个较大的失重峰,此时TG-MS 中只有CO2的信号,认为这是Cu-Mn 复合碳酸盐分解产生的。这种复合盐可能来源于两方面:一方面是由于样品的少量陈化(XRD 显示存在少量结晶)形成了少量的复合碳酸盐;另一方面,考虑到这一阶段的失重率较大,也存在热分解过程中形成了部分复合碳酸盐的可能。由于微反应器优异的混合效果,沉淀物中Cu-Mn 分布得非常均匀,这对热分解过程中形成复合碳酸盐是有利的。

图5 沉淀物的TG-MS曲线Fig.5 TG-MS curves of the precipitates
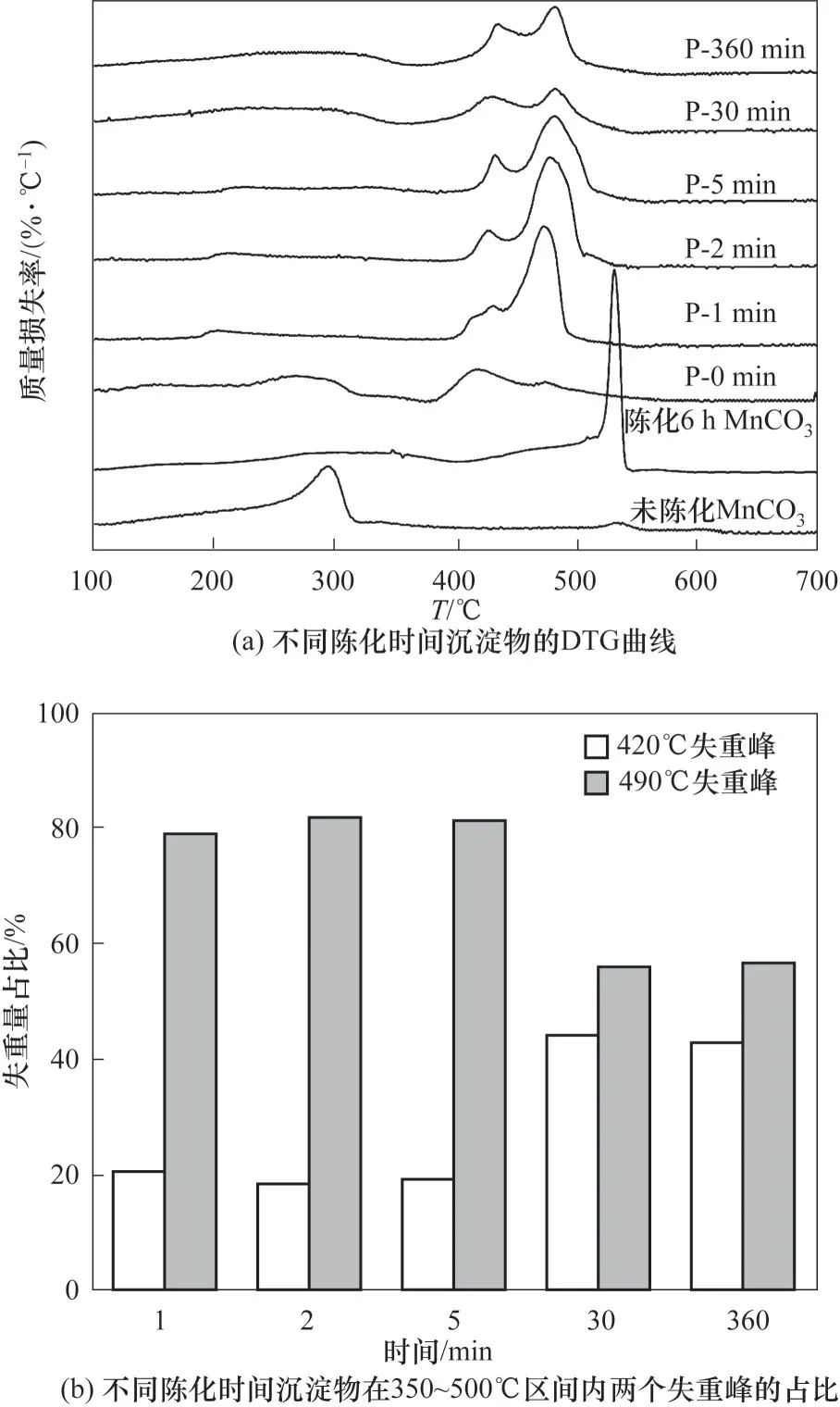
图6 沉淀物的TGA分析Fig.6 TGA analysis of the precipitates
陈化后样品的热分解行为发生了明显变化:一方面,TG-MS 中H2O 信号强度快速减弱至完全消失,表明碱式碳酸盐在陈化过程中快速转化为碳酸盐;另一方面,热分解过程主要发生在400~500℃区间,而失重峰的形状随着陈化时间的增加逐渐发生变化。对于长时间陈化的样品P-360 min,在420 和490℃出现两个失重率相近的峰,TG-MS 分析表明两者都是碳酸盐的分解。XRD 分析表明陈化后的样品中存在着结晶态的MnCO3,文献中报道MnCO3分解温度高于CuCO3[12,16],故认为490℃的失重峰对应于MnCO3的分解。相较于单纯MnCO3陈化后较高的分解温度[见图6(a)中陈化6 h 的MnCO3曲线],由于Cu 的存在,P-360 min 中MnCO3分解温度显著下降。根据前文XRD 分析中样品MnCO3晶粒尺寸小于纯MnCO3的结论,推测是小晶粒的不稳定性使其在较低温度分解。而对于420℃失重峰,鉴于其对应的失重率大于单纯CuCO3分解能够产生的失重率,认为该失重峰对应于Cu-Mn 复合碳酸盐的分解。
对于陈化时间从1~30 min 的样品,其DTG 曲线的变化体现了无定形的沉淀物逐步演变到结晶态陈化产物的过程:首先是结晶态的MnCO3快速形成,490℃的失重峰在陈化5 min 的样品中已经非常完整;其次是Cu-Mn 复合碳酸盐的缓慢形成,与其对应的420℃左右的分解峰所占比例在陈化5 min以后逐步提升,峰形逐渐完整。图6(b)为这两个失重峰的相对比例关系,表明420℃左右的失重率从20%上升至40%左右。
根据上述分析,认为未陈化样品主要由无定形碱式碳酸铜和MnCO3组成,长时间陈化后样品主要由结晶态的MnCO3和Cu-Mn 复合碳酸盐组成。陈化过程包含了2快1慢3个过程:无定形MnCO3快速转变为结晶态的过程;碱式碳酸铜在Mn 存在的情况下迅速脱水的过程;Cu2+逐渐进入结晶态的MnCO3结构中,缓慢形成Cu-Mn 复合碳酸盐的过程。在陈化开始阶段的结构快速变化过程中,MnCO3转变为结晶态,导致微观层面上Cu、Mn 的分离;随着陈化的进行,Cu-Mn 复合碳酸盐的形成使得Cu、Mn 的相互分散均匀性得以提高。陈化过程中这些变化可以用图7 简略表示:Cu、Mn 为处于良好分散状态的无定形沉淀物,它们不同的结晶速率导致陈化初期(1~5 min)的Cu、Mn分离,在热力学驱动力作用下Cu-Mn 复合碳酸盐形成,提高了两者的分散性。

图7 陈化过程沉淀物结构变化示意图Fig.7 Schematic diagram of structural changes of precipitates during aging
从热分解过程和前面的分析可以看出,Cu、Mn之间存在着较强的相互作用,在陈化过程中互相作用于对方的结构演变过程。一方面,Mn 的存在使Cu 无法形成稳定的孔雀石结构(结晶态碱式碳酸铜);另一方面,Cu 的存在不仅降低了MnCO3的分解温度,而且还会形成Cu-Mn 复合碳酸盐。显然,这种相互作用会体现在催化性能上:Cu-Mn 复合碳酸盐焙烧后得到的复合氧化物催化性能更佳[12];Cu 的存在大幅降低了沉淀物整体的分解温度,400℃附近沉淀物已基本分解,减少了烧结,有利于催化活性的提高[11,17-18]。因此,陈化过程中的结构变化非常好地说明了催化剂活性的变化过程:未陈化的沉淀物中Cu-Mn 的混合非常均匀,具有很好的催化活性;短时间陈化(5 min 左右)后,Cu、Mn 相互分离,催化剂的活性最差;随后经过长时间陈化,Cu-Mn 复合碳酸盐形成,催化活性又逐步恢复。由此可见,Cu-Mn 元素分散性对催化剂活性有着重要的影响。未陈化样品和陈化30 min 以后的样品虽然在结晶度上存在着明显的不同,但两者共同具有的良好的Cu-Mn 相互分散性,使其焙烧后得到的催化剂活性均较好。
2.4 陈化过程对催化剂的影响
陈化过程中Cu-Mn 分散性的变化是通过催化剂结构作用于催化性能,下面对催化剂结构进行研究。不同焙烧温度的实验表明,410℃下焙烧2 h 得到的催化剂具有最好的活性。对其进行XRD 分析(图8)可以看到,所有样品中都没有看到Cu、Mn 氧化物的相关特征峰。根据文献[17-18]可知,当焙烧温度在400℃附近时Cu-Mn 催化剂只能形成无定形复合氧化物结构,因而不会在XRD 谱图中出峰。对比PDF标准卡片(PDF#01-083-1763)可以发现,陈化较短时间的样品(C-1 min~C-5 min)在31°和52°附近出现了MnCO3(104)和(116)晶面特征峰,即存在未分解的MnCO3。这说明短时间陈化的样品主要是结晶态MnCO3,其分解温度较高。陈化时间延长后,Cu 进入MnCO3晶格形成Cu-Mn 复合碳酸盐,其在410℃下焙烧能够分解,这样导致残留的MnCO3数量较少,XRD谱图中不再出峰。

图8 不同陈化时间催化剂的XRD谱图Fig.8 XRD patterns of catalysts with different aging time
为得到更多信息,对催化剂样品进行了拉曼分析,如图9 所示。可以看到,不同催化剂样品均在630~640 cm-1范围内出现了明显的拉曼峰。参考相关文献[19-23]可知,MnOx中Mn-O-Mn 的出峰位置在644 cm-1左右,当存在Cu组分时,Cu、Mn间由于相互作用会生成Mn-O-Cu,使样品原本644 cm-1左右的拉曼出峰向低波数偏移[20,23],且Cu-Mn 相互作用越强时偏移量越大[20]。样品C-0 min~C-360 min 的出峰位置分别为632、634、640、636 和634 cm-1,拉曼峰的偏移量先变小后变大,说明Cu-Mn 相互作用先减弱后增强。对比催化性能变化规律可以发现,Cu-Mn 相互作用的变化规律与其相符,说明较强的Cu-Mn 相互作用有利于催化效果提高,这与文献中结果一致[24]。同时,结合前文陈化过程的分析,认为随着陈化的进行,Cu-Mn 元素均匀性先变差再变好的过程是催化剂中Cu-Mn 相互作用变化的原因,即沉淀物中Cu-Mn 元素的均匀性更好会导致催化剂中更强的Cu-Mn 相互作用,并最终影响了催化剂性能。

图9 不同陈化时间催化剂的拉曼谱图Fig.9 Raman spectra of catalysts with different aging time
催化剂的表面性质是影响其性能的一个重要因素,通过XPS 表征考察不同陈化时间对催化剂表面性质的影响。图10(a)为Mn 2p的XPS谱图,其中结 合 能 为638~648 eV 的Mn 2p3∕2轨 道 峰 可 以 分 为Mn3+、Mn4+[25-27]两部分,结合能分别处于641.8 和643.5 eV 附近。经过分峰拟合可以发现,Mn4+峰先增大后减小的趋势较为明显。图10(b)为不同催化剂样品的O 1s 谱图,其XPS 峰可以分为529~530、530~532 和533~534 eV 三部分,分别对应晶格氧(Olatt)、吸附氧(Oads)和吸附的OH-或水分子中的氧[28-29]。分峰拟合表明,晶格氧对应的峰先减小后增大,而吸附氧对应峰先增大后减小。图10(c)为催化剂Cu 2p 的XPS 谱图,其中629~637 eV 的Cu 2p3∕2轨道峰可以分为Cu+和Cu2+两部分,结合能分别处于931.0和933.8 eV 附近[28,30]。同样由分峰拟合得到Cu+的峰先减小后增大,而Cu2+的峰先增大后减小。
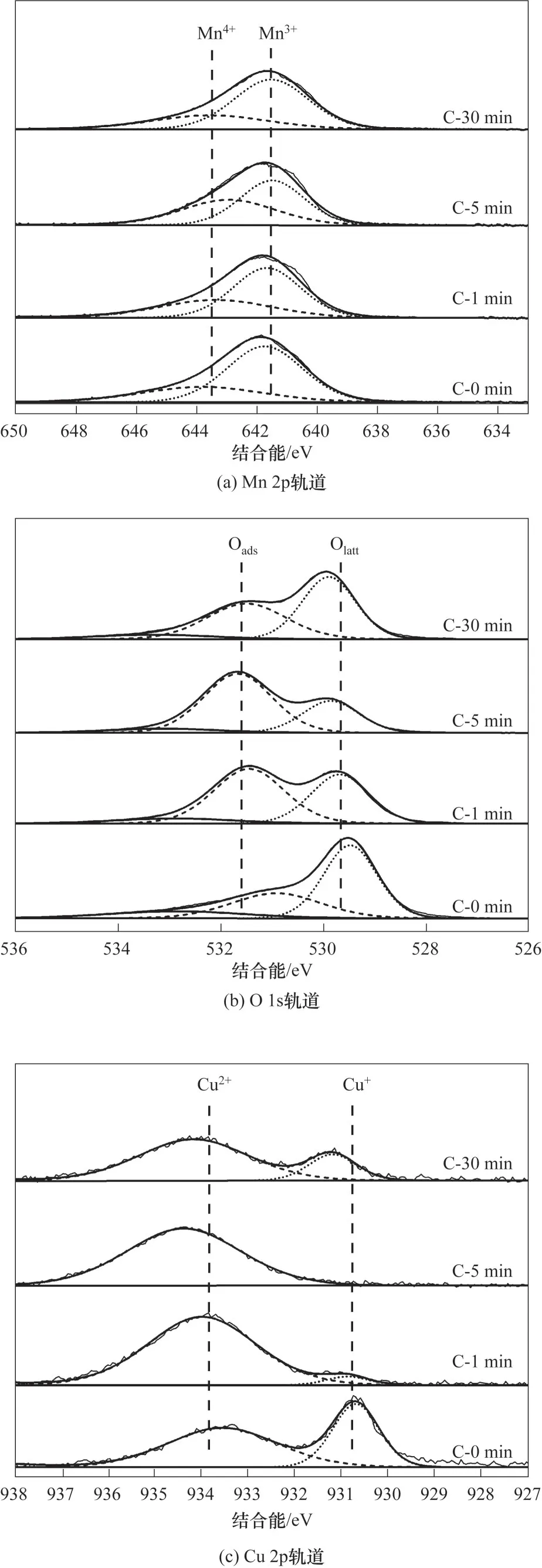
图10 不同陈化时间催化剂的XPS谱图Fig.10 XPS spectra of catalysts with different aging time
为使实验结果更直观,将不同陈化时间的Cu-Mn 催化剂表面物种变化用图11 表示。可以看到,随着陈化时间的延长,样品表面Mn3+∕Mn4+出现了先减小后增大的现象并在C-5 min 样品处存在极值;Cu+∕Cu2+整体呈现先下降后上升的趋势,在C-5 min时达到最小值,此时几乎不存在Cu+;而Olatt∕Oads总体呈下降趋势。文献指出,催化剂的表面氧物种和Cu、Mn 元素的氧化态等均对催化活性有影响[1]。当Cu和Mn处于较低氧化态时,金属由于电子密度高,容易向吸附氧提供电子,形成更多的活性氧离子从而对催化剂的活性有利[28]。Huang 等[19]研究发现Mn3+有利于Olatt在催化剂表面的迁移,可以提高Olatt的活性,并且Mn3+的出现往往伴随着氧空位的出现,更多的氧空位也有利于催化活性,故C-0 min 样品最高的Mn3+∕Mn4+、Cu+∕Cu2+及晶格氧比率可能是它催化活性优异的一方面原因。同时,Mn3+和Cu+比率先变低至陈化5 min 后再变高最终趋于稳定的过程,与沉淀物中Cu-Mn 元素分散性变化规律相似,故推测表面Cu、Mn 价态的变化本质上也来源于陈化过程中Cu-Mn 之间的相互作用,这最终导致了催化剂性能呈现先迅速变差至5 min 后逐渐变好最终达到稳定的变化规律。

图11 不同陈化时间催化剂表面物种变化趋势Fig.11 Surface species change trend of catalysts at different aging time
催化剂结构表征显示,Cu-Mn 元素相互作用、金属元素的价态分布等特性均与陈化物中Cu-Mn的相互分散性相关,并与催化性能的变化趋势一致。这一结果不仅支持了陈化过程中Cu-Mn 分散性的变化规律,也为探索陈化过程作用于催化性能的机制提供了依据。
3 结 论
本文利用微反应器进行Cu-Mn 共沉淀,将沉淀物分别经过不同时间的陈化,研究陈化过程对Cu-Mn沉淀物结构及其后续制备的催化剂的影响,得到如下结论。
(1)在陈化过程中,共沉淀产物中均匀分布的Cu 和Mn 由于结晶态MnCO3的快速形成而相互分离,随后Cu-Mn复合碳酸盐的形成使得Cu-Mn的分散性再次变好。陈化过程中Cu-Mn 分散性的变化规律,与催化剂活性随陈化时间增加先迅速下降后上升的趋势一致。
(2)由于微反应器较好的混合效果,并能最大限度地避免制备过程中的部分陈化,因此得到的初始沉淀产物中Cu-Mn 元素具有极好的分散性,其焙烧得到的催化剂活性高,且优于长时间陈化后得到的催化剂。
(3)无定形MnCO3转变为结晶态是一个快速过程,在数分钟内即能完成;而Cu2+进入结晶态MnCO3结构中是一个慢过程,可持续数十分钟。陈化开始后,随着结晶态MnCO3的形成,Cu-Mn 元素分散性迅速变差;长时间陈化后,随着Cu-Mn 复合碳酸盐的形成,Cu-Mn分散性又逐渐变好,并最终在30 min左右达到稳定状态。
(4)催化剂表征显示,不同陈化时间样品中的Cu-Mn 分散性与最终催化剂中Cu-Mn 元素相互作用强弱有关,也与催化剂表面Mn3+∕Mn4+、Cu+∕Cu2+比例的变化趋势相同,最终决定了催化剂的活性。
猜你喜欢
农家参谋(2021年5期)2021-06-20
重庆理工大学学报(自然科学)(2020年12期)2021-01-21
大众科学(2020年8期)2020-11-02
中国化工贸易·上旬刊(2018年9期)2018-09-10
阅读(科学探秘)(2018年10期)2018-05-14
求知导刊(2017年14期)2017-07-15
科技资讯(2017年13期)2017-06-21
小学生·新读写(2014年8期)2014-10-21
