
α-羰酰氧基苯乙酮及α-羟基苯乙酮的制备
2020-09-09 06:48:44陈小文黄志诚宋祥家任红霞王立新
合成化学 2020年8期
陈小文,黄志诚 ,邹 滢,李 霞,宋祥家,任红霞,田 芳,王立新
(1.中国科学院 成都有机化学研究所,四川 成都 610041;2.中国科学院大学,北京 100049)
α-羟基酮骨架是各种生物活性化合物和天然产物的特征结构[1-5]与合成前体[6-9]。一般通过下列方法制备:1)在微波[10]或高压汞灯[11]辐射下,α-溴代酮直接发生羟基取代反应;2)在过氧化钼[12]或其他氧化剂[13]作用下烯醇发生α-羟基化作用;3)通过分子氧氧化烯胺衍生物[14];4)金属催化氧化烯烃[15];5)采用间氯过氧苯甲酸[16]或类似氧化剂[17]使烯醇硅醚发生氧化。
Ismail等[18]采用一锅法,苯基环氧乙烷化合物溶于四氢呋喃中,在(NH4)6Mo3O24·4H2O/H2O2体系中发生氧化开环反应,生成羟基苯乙酮,收率94%。该方法使用的催化剂用量(1 eq.)较大,不利于成本控制。Zhang等[19]在廉价12-钼磷酸和十六烷基吡啶氯化物体系的催化作用下,苯乙烯与过氧化氢直接发生酮羟基化,以良好的收率和优异的选择性得到羟基苯乙酮化合物,收率为86.4%。但是只有提高催化剂用量,反应才能发生,而且催化剂活性随着使用次数的增加而降低。Utsukihara等[20]采用微波辅助的方法,在氨水溶液中,邻溴环己酮发生羟基化反应转变为邻羟基环己酮,收率仅24%。在生成产物的同时,邻溴环己酮也会与氨水发生缩合反应生成吡嗪衍生物以及脱溴生成环己烯酮等副产物,导致产物分离困难,收率降低。Wong等[21]在甲酸铯作用下,溴代苯乙酮在无水甲醇溶剂中转变为羟基苯乙酮,转化率可达98%,但该反应使用的原料甲酸铯价格昂贵,其他甲酸盐的效果并不太理想,在使用氢氧化钠等常用廉价碱的情况下,收率明显降低(约51%)。
为了探索制备α-羟基苯乙酮的新方法,本文在季铵盐催化下,溴代苯乙酮(1)与醋酸钠(2)反应得α-乙酰氧基苯乙酮(3),最后在碱性条件下高收率制得α-羟基苯乙酮(4,Scheme1),其结构经1H NMR确证。

Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker-300 MHz型核磁共振仪(TMS为内标)。
所用试剂均为分析纯。
1.2 合成
(1)3的合成
将1(2.5 mmol)、2(5.0 mmol)、四丁基溴化铵(TBAB)(0.10 mmol)加入乙腈4.0 mL中,回流反应2.0 h(TLC监测,展开剂:PE/EA=10/1,V/V)。过滤,滤液减压蒸馏至2~3 mL,加入50%乙醇-水溶液2.0 mL,置于冰箱中冷却,过滤,滤液用50%乙醇-水溶液洗涤,真空干燥得3383.2 mg,纯度98.6%(HPLC);1H NMR(300 MHz,CDCl3)δ:7.90~7.87(m,2H),7.60~7.55(m,1H),7.48~7.43(t,2H),5.31(s,2H),2.20(s,3H)。
(2)4的合成
将3(1.0 mmol)加入到甲醇2.0 mL中,缓慢滴加0.2 N氢氧化钠溶液5.0 mL,滴毕,反应3.0 h。减压蒸除溶剂,残余物加入2-丁酮10 mL,煮洗,过滤得白色固体醋酸钠94.6 mg,可重复用于3的合成,滤液减压蒸除溶剂得黄色固体4115.2 mg,纯度95.8%;1H NMR(CDCl3)δ:7.93~7.90(q,2H),7.65~7.60(m,1H),7.52~7.47(q,2H),4.87(s,2H),3.50(s,1H)。
表1为合成3反应的条件优化。由表1可知,最佳反应条件为:3%mmol TBAB为催化剂,乙腈为溶剂,在80 ℃下反应2.0 h。在最佳反应条件下,可以以100.0%的转化率和99.2%的选择性得到乙酰氧基苯乙酮。
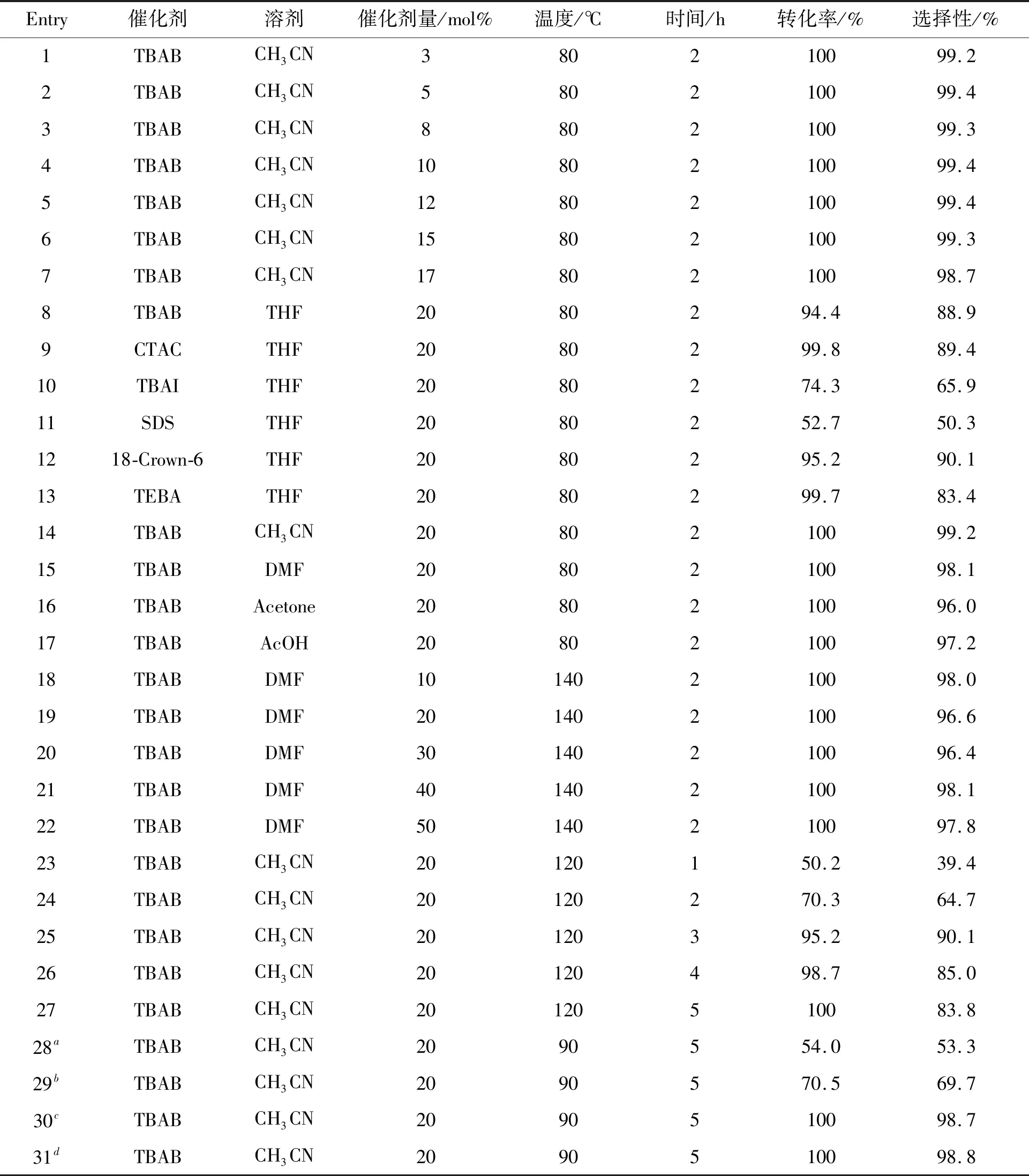
表1 反应条件的筛选Table 1 Screenings of the reaction conditions
表2为羰酰氧基苯乙酮底物的扩展。由表2可知,溴代苯乙酮制备羰酰氧基苯乙酮的反应具有良好的普适性,产物收率均达到良好至优异。
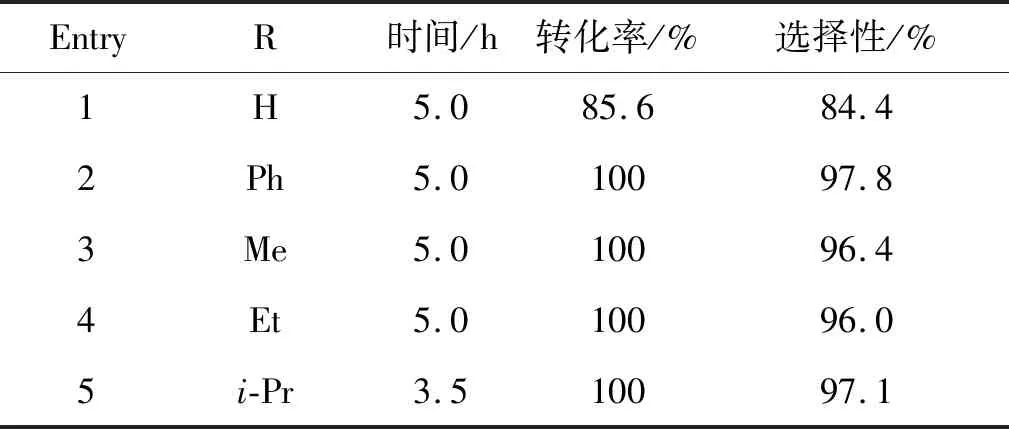
表2 羰酰氧基苯乙酮底物的扩展Table 2 Generality of carboxyloxy acetophenone
表3为使用回收醋酸钠制备α-乙酰氧基苯乙酮的反应结果。由表3可知,使用回收的醋酸钠参与反应,反应转化率可达100%,纯度为97.6%,选择性97.5%~99.0%。
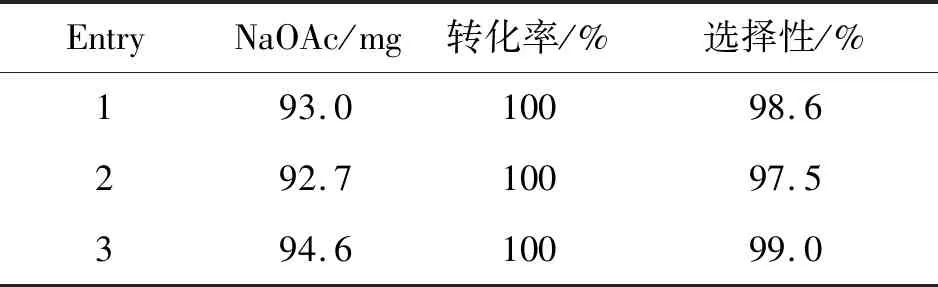
表3 使用回收醋酸钠制备α-乙酰氧基苯乙酮Table 3 Preparation of acetoxy acetophenone using recycled sodium acetate
报道了一种合成α-羟基苯乙酮的新方法。该方法具有成本低廉、反应高效迅速、后处理方便、副反应少等优点,具有潜在的工业应用前景。
猜你喜欢
当代化工研究(2021年22期)2021-04-11 18:12:22
上海计量测试(2020年6期)2021-01-15 03:18:14
中国化工贸易·上旬刊(2020年3期)2020-09-10 07:22:44
消费导刊(2019年14期)2019-08-21 01:00:51
消费导刊(2019年27期)2019-07-22 09:12:22
CHINESE JOURNAL OF AERONAUTICS(2017年1期)2017-11-21 12:54:14
合成化学(2015年2期)2016-01-17 09:03:25
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
中国病理生理杂志(2015年8期)2015-12-21 12:38:14
化学教与学(2012年6期)2012-03-20 14:05:57
