
新型5-取代苯基的呋喃类化合物的合成
2020-09-09 07:18张泽琴陈美玲肖若彤
合成化学 2020年8期
张泽琴,陈美玲,肖若彤,洪 伟
(北方民族大学 化学与化学工程学院,宁夏 银川 750021)
呋喃环结构广泛地存在于药物和天然产物的结构中[1-4],Lorente等[5-6]曾经报道了含有呋喃环的天然产物在抗癌、抗炎、抗感染、免疫抑制、抗氧化等方面展现了较好活性。将呋喃环结构引入母体化合物,可有效改善其生物活性[7-8]。呋喃环作为新药的候选物,近年来需求不断[9-10],因此合成出以呋喃环为母核的系列衍生物具有重要现实意义。
本文报道的新化合物5a~5e是在呋喃环的2-位引入Boc保护的丙二胺基团侧链,5-位引入不同取代的苯环,这一系列化合物的特点是5-位引入的取代的苯环可以进行进一步的结构改造。以N,N′-二甲基丙二胺(1)为原料,通过Boc保护反应制备得到N-叔丁氧羰基-N,N′-二甲基丙二胺(2);再与5-溴糠醛(3)在三乙酰氧基硼氢化钠作为还原剂的作用下进行还原胺化制备得到N-叔丁氧羰基-N′-(5-溴呋喃-2-基-甲基)-N,N′-二甲基丙二胺(4);最后以1,1′-二叔丁基膦基二茂铁二氯化钯作为催化剂与一系列不同取代的苯硼酸进行Suzuki反应合成化合物5a~5e(Scheme 1),其结构经1H NMR和HR-MS(ESI)表征。
1 实验部分
1.1 仪器与试剂
Bruker Avance 400 MHz型核磁共振仪(TMS为内标);FTIR-1500型红外光谱仪(KBr压片);Accurate-Mass O-TOF LC/MS型质谱仪。
所用试剂均为分析纯。
1.2 合成
(1)2的合成
在干燥的三口瓶中加入N,N′-二甲基丙二胺(1)5.00 g(48.90 mmol)和甲醇30 mL,搅拌使其溶解;缓慢滴加二碳酸二叔丁酯3.56 g(16.30 mmol)的(15 mL)二氯甲烷溶液,滴毕,冰盐浴冷却,于20 ℃反应48 h。升温至室温,减压浓缩除去溶剂,残余物加入NaHCO3饱和溶液50 mL,用DCM(3×30 mL)萃取,合并有机相,经无水硫酸钠干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂:V(MeOH)/V(DCM)=1/50]纯化,浓缩得淡黄色油状液体22.45 g,收率74%;1H NMR(CDCl3,400 MHz)δ:3.28(d,J=8.6 Hz,2H,CH2),2.84(s,3H,CH3),2.56(t,J=6.9 Hz,2H,CH2),2.44(d,J=9.1 Hz,2H,CH2),2.01(s,3H,CH3),1.46(s,9H,CH3)。
(2)4的合成
在干燥的三口瓶中加入化合物23.69 g(18.25 mmol)和1,2-二氯乙烷60 mL ,搅拌使其溶解;加入化合物32.20 g(12.17 mmol),于25 ℃反应1 h。溶液变为棕褐色,加入三乙酰氧基硼化钠5.20 g(24.52 mmol)和1,2-二氯乙烷40 mL(各分两批加入,间隔6 h),反应24 h。加入氯化铵饱和溶液50 mL淬灭反应,减压浓缩除去溶剂,残余物用DCM(3×30 mL)萃取,合并有机相,用无水Na2SO4干燥,浓缩,残余物经硅胶柱层析[洗脱剂:V(EA)/V(PE)=1/3]纯化,浓缩得黄色油状液体43.45 g,收率79%;1H NMR(DMSO-d6,400 MHz)δ:6.48(d,J=3.2 Hz,1H,ArH),6.34(d,J=3.2 Hz,1H,ArH),3.37(s,2H,CH2),3.15(t,J=7.1 Hz,2H,CH2),2.75(d,J=4.9 Hz,3H,CH3),2.26(t,J=7.1 Hz,CH2),2.13(s,3H,CH3),1.61(p,J=6.7,6.0 Hz,2H,CH2),1.38(s,9H,CH3);FT-IRν:2933.23,1694.20,1365.23,1307.69,1164.68,1126.37,1047.52,1013.87 cm-1。
(3)5a~5e的合成通法
在干燥的三口瓶中加入化合物41.39 mmol和甲苯/水/乙醇(2/1/1)的混合溶剂20 mL,搅拌使其溶解;依次加入取代的苯硼酸2.09 mmol,碳酸钾4.17 mmol和催化剂1,1′-二叔丁基膦基二茂铁二氯化钯0.03 mmol,于80 ℃回流反应8 h。加入NaHCO3饱和溶液10 mL,EA(3×10 mL)萃取,合并有机相,用无水Na2SO4干燥,过滤,浓缩,残余物经硅胶柱层析[洗脱剂:V(EA)/V(PE)=1/2]纯化,浓缩得化合物5a~5e。
N-叔丁氧羰基-N′-(5-(4-氰基苯基)呋喃-2-基-甲基)-N,N′-二甲基丙二胺(5a):黄色油状液体0.43 g,收率81%;1H NMR(DMSO-d6,400 MHz)δ:7.85(q,J=8.3 Hz,4H,ArH),7.18(d,J=3.4 Hz,1H,ArH),6.48(d,J=3.4 Hz,1H,ArH),3.61(s,2H,CH2),3.17(t,J=7.1 Hz,2H,CH2),2.76(s,3H,CH3),2.33(t,J=7.0 Hz,2H,CH2),2.20(s,3H,CH3),1.65(t,J=7.4 Hz,2H,CH2),1.36(s,9H,CH3);FT-IRν:2348.81,2224.59,1690.69,1307.24,1160.28,1059.70,662.55 cm-1;HR-MS(ESI-TOF)m/z:Calcd for C22H30N3O3+{[M+H]+}384.2293,found 384.2287。
N-叔丁氧羰基-N′-(5-(3-氰基苯基)呋喃-2-基-甲基)-N,N′-二甲基丙二胺(5b):黄色油状液体0.42 g,收率79%;1H NMR(DMSO-d6,400 MHz)δ:8.12(t,J=1.7 Hz,1H,ArH),7.98(d,J=7.9 Hz,1H,ArH),7.73(dt,J=7.7,1.4 Hz,1H,ArH),7.63(t,J=7.8 Hz,1H,ArH),7.12(d,J=3.3 Hz,1H,ArH),6.45(d,J=3.3 Hz,1H,ArH),3.60(s,2H,CH2),3.17(t,J=7.1 Hz,2H,CH2),2.76(s,3H,CH3),2.32(t,J=7.2 Hz,2H,CH2),2.20(s,3H,CH3),1.65(s,2H,CH2),1.36(s,9H,CH3);FT-IRν:2229.75,1692.34,1304.80,1158.86,1023.40,685.73 cm-1;HR-MS(ESI-TOF)m/z:Calcd for C22H30N3O3+{[M+H]+}384.2292,found 384.2287。
N-叔丁氧羰基-N′-(5-(4-甲氧碳基苯基)呋喃-2-基-甲基)-N,N′-二甲基丙二胺(5c):黄色油状液体0.46 g,收率79%;1H NMR(DMSO-d6,400 MHz)δ:8.06~7.94(m,2H,ArH),7.80(d,J=8.1 Hz,2H,ArH),7.10(d,J=3.3 Hz,1H,ArH),6.46(d,J=3.3 Hz,1H,ArH),3.60(s,2H,CH2),3.17(t,J=7.2 Hz,2H,CH2),2.76(s,3H,CH3),2.33(t,J=7.0 Hz,2H,CH2),2.20(s,3H,CH3),1.65(m,2H,CH2),1.36 (s,9H,CH3);FT-IRν:1692.91,1638.67,1276.62,1176.85,1018.55,770.94 cm-1;HR-MS(ESI-TOF)m/z:Calcd for C23H33N2O5+{[M+H]+}417.2382,found 417.2389。
N-叔丁氧羰基-N′-(5-(4-硝基苯基)呋喃-2-基-甲基)-N,N′-二甲基丙二胺(5d):黄色油状液体0.51 g,收率92%;1H NMR(DMSO-d6,400 MHz)δ:8.31~8.25(m,2H,ArH),7.91(d,J=8.6 Hz,2H,ArH),7.27(d,J=3.3 Hz,1H,ArH),6.52(d,J=3.4 Hz,1H,ArH),3.36(s,2H,CH2),3.18(t,J=7.1 Hz,2H,CH2),2.76(s,3H,CH3),2.33(t,J=7.1 Hz,2H,CH2),2.21(s,3H,CH3),1.64(d,J=10.7 Hz,2H,CH2),1.37(d,J=6.7 Hz,9H,CH3);FT-IRν:1692.17,1514.54,1333.37,1159.13,1023.93,851.98 cm-1;HR-MS(ESI-TOF)m/z:C21H30N3O5+{[M+H]+}404.2191,found 404.2185。
N-叔丁氧羰基-N′-(5-(3-硝基苯基)呋喃-2-基-甲基)-N,N′-二甲基丙二胺(5e):黄色油状液体0.45 g,收率80%;1H NMR(DMSO-d6,400 MHz)δ:8.41(t,J=2.0 Hz,1H,ArH),8.12(dd,J=8.2 Hz,2.2 Hz,2H,ArH),7.72(t,J=8.0 Hz,1H,ArH),7.21(d,J=3.3 Hz,1H,ArH),6.48(s,1H,ArH),3.63(s,2H,CH2),3.18(t,J=7.1 Hz,2H,CH2),2.76(s,3H,CH3),2.35(m,2H,CH2),2.22(s,3H,CH3),1.66(m,2H,CH2),1.36(s,9H,CH3);FT-IRν:1692.80,1349.42,1305.36,1159.36,1060.18,862.71 cm-1;HR-MS(ESI-TOF)m/z:Calcd for C21H30N3O5+{[M+H]+}404.2192,found 404.2185。
在化合物2的制备过程中,根据文献[11-12]方法,使用二碳酸二叔丁酯对N,N′-二甲基丙二胺进行单保护反应。此步骤中,加料的顺序和速度非常重要。为避免双保护的副产物生成,将二碳酸二叔丁酯的二氯甲烷溶解溶液置于滴液漏斗中,控制滴加速度,缓慢滴至N,N′-二甲基丙二胺的甲醇溶液中。此外,还探索了N,N′-二甲基丙二胺与二碳酸二叔丁酯的摩尔比分别为2/1和3/1的条件下,化合物2的收率变化。结果表明,当N,N′-二甲基丙二胺与二碳酸二叔丁酯的摩尔比为2/1时,化合物2的收率52%,而当摩尔比为3/1时,收率74%。因此确定N,N′-二甲基丙二胺与二碳酸二叔丁酯的摩尔比为3/1。
根据还原胺化反应[13],将2与3进行制备得到4。反应过程中2的氨基先与3的醛基反应生成亚胺,再被还原剂还原得到叔胺,因此中间体亚胺的生成时间较长。还原剂选用三乙酰氧基硼氢化钠[14],是因为化合物3的结构中有醛基,而三乙酰氧基硼氢化钠作为还原剂对亚胺具有高度的选择性,在反应的体系中不会把醛基还原成羟基而导致影响收率。而此反应体系中的溶剂选用1,2-二氯乙烷[15],是因为文献[16]证实1,2-二氯乙烷对醛类、胺类、还原剂有较好的溶解性。另外,探索了化合物3与三乙酰氧基硼氢化钠在不同摩尔比分别为1/1,1/1.4,1/1.8,1/2的条件下,化合物4收率的变化情况(表1)。结果表明,n(3)/n(三乙酰氧基硼氢化钠)为1/2时,收率最高(79%)。此外,采用分批加入的方法可避免三乙酰氧基硼氢化钠长时间与氧气接触导致的变质。

表1 不同的摩尔比中化合物4收率的变化情况Table 1 Changes in the yield of compound 4 in different molar ratios
本课题组之前报道过,用催化剂四(三苯基磷)钯在微波辅助下,经Suzuki反应合成类似化合物的路线[17]。在非微波条件下[18-19],使用四(三苯基磷)钯催化Suzuki反应[20-21],反应时间较长且收率较低。如果采用1,1′-二叔丁基膦基二茂铁二氯化钯作催化剂进行Suzuki反应,在非微波条件下,反应时间也较短,收率较高。
合成化合物5a~5e的过程中,采用1,1′-二叔丁基膦基二茂铁二氯化钯为催化剂,无水碳酸钾为碱,反应时间为8 h,比较了两种溶剂体系{V1[V(甲苯)/V(乙醇)/V(水)=2/1/1)]与V2[(V(四氢呋喃)/V(水)=1/1]为溶剂}对化合物5a~5e收率的影响(表2)。由表2可知,溶剂为V1时,化合物5a~5e的收率普遍偏高,溶剂为V2时,化合物5a~5e的收率较低。
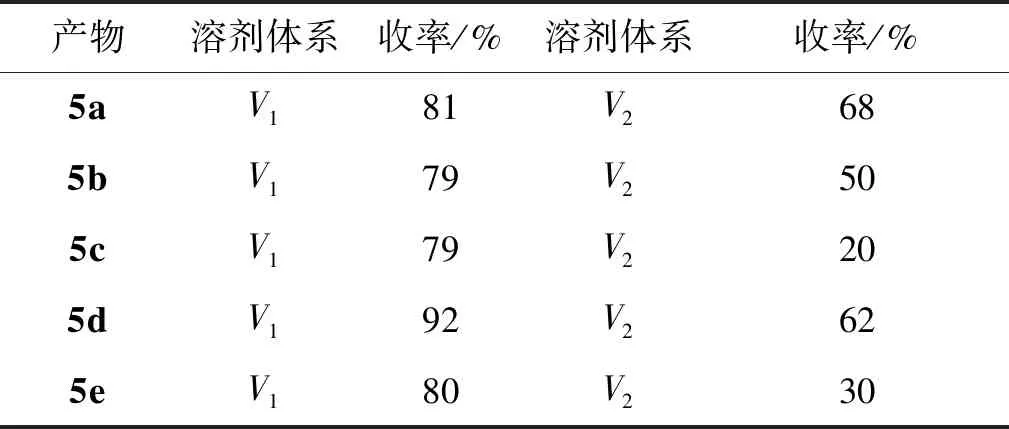
表2 不同溶剂中化合物5a~5e收率的比较Table 2 Comparison of compound yields of 5a~5e in different solvents
以N,N′-二甲基丙二胺为原料进行单Boc保护反应制得N,N′-二甲基-N-叔丁氧羰基丙二胺;再与5-溴糠醛在三乙酰氧基硼氢化钠的作用下发生还原胺化得到中间体N,N′-二甲基-N-叔丁氧羰基-(5-溴呋喃-2-基-甲基)-丙二胺;此化合物在新型催化剂1,1′-二叔丁基膦基二茂铁二氯化钯的作用下与一系列苯硼酸发生Suzuki反应,合成了目标化合物5a~5e。
猜你喜欢
世界农药(2021年3期)2021-12-09
浙江大学学报(理学版)(2021年5期)2021-09-17
食品安全导刊(2021年20期)2021-08-30
上海理工大学学报(2021年3期)2021-07-20
河南化工(2021年3期)2021-01-08
合成生物学(2020年1期)2020-07-15
食品与发酵工业(2020年12期)2020-07-07
中国食品(2020年9期)2020-05-26
农药科学与管理(2019年8期)2019-11-23
分析化学(2017年6期)2017-06-15
