
PPARs在不同类型心力衰竭中作用的研究进展*
2020-09-07 09:36张健维
中国病理生理杂志 2020年8期
张健维,徐 坦,徐 明
(北京大学第三医院心血管内科、血管医学研究所,心血管分子生物学与调节肽卫计委重点实验室,分子心血管学教育部重点实验室,北京100191)
过氧化物酶体增殖物激活受体(peroxisome pro‑liferator-activated receptors,PPARs)是1990年发现的核受体超家族的成员,迄今为止共发现了3 种PPARs(α、β/δ 和 γ)。PPARs 主要在肝、脂肪、心肌等组织表达,发挥促进脂肪酸摄取、转运和氧化,参与能量代谢平衡的功能[1]。心力衰竭(简称“心衰”)时心脏重塑的机制与能量稳态直接相关[2],随着糖尿病、高血压等代谢性疾病所致慢性心衰的发病率逐年上升,代谢调控在心衰治疗中的作用受到了广泛关注[3-4]。
1 PPARs的结构和功能
PPARs 由N/C 末端配体结合结构域和DNA 结合结构域构成,见图1[5]。PPARs 被激活后,与类视黄醇X 受体(retinoid X receptor,RXR)异二聚化结合,并锚定至靶基因启动子区域特异性DNA 序列——过氧化物酶体增殖物反应元件(peroxisome prolifera‑tor response element,PPRE),协同增强靶基因转录,调控下游基因表达。PPARγ 辅激活因子1α(PPARγ coactivator-1α,PGC-1α)是其经典辅激活因子,细胞外信号调节激酶(extracellular signal-regulated ki‑nase,ERK)等可调控PPARs 活性。PPARs 的配体包括PPARα 激动剂贝特类和PPARγ 激动剂格列酮类药物,脂肪酸亦对其具有激活作用。
PPARs 在不同病因诱导的心衰中发挥不同的调控作用。以下将从高血压性心衰、缺血性心衰和糖尿病性心衰3 种类型分别论述PPARs 相关效应机制和调控靶点。
2 PPARs与高血压性心衰

Figure 1.Various structures of the subtypes of PPARs [5].AF-1/2:activation function-1/2;DBD:DNA-binding domain;LBD:li‑gand-binding domain.图1 PPARs亚型的不同结构
高血压性心衰的重要机制是与心肌细胞相匹配的微循环能量供应缺乏,而心肌细胞能量代谢模式的变化,即代谢重塑(metabolic remodeling),又将加剧心肌细胞的氧化应激损伤,最终导致心肌细胞凋亡[6-7]。在生理情况下和压力负荷心衰早期,PPARs可上调脂代谢基因表达,以维持心肌以脂代谢为主的能量供应方式[8]。随着心衰的进展,PPARs 表达下调,脂肪酸氧化功能降低,代谢底物向葡萄糖转变;发展至晚期,心脏负荷增加且缺氧加剧,导致绝大多数葡萄糖经无氧糖酵解转化为乳酸,其能量效率大幅减低,从而导致能量缺乏,并引发线粒体功能障碍、氧化应激,发生心肌重塑[9]。PPARα、PPARγ和PPARβ/δ不同亚型各具特点。
2.1 PPARα 促进心肌脂代谢 PPARα 的激活可上调脂肪酸氧化途径关键酶,促进心肌细胞β 氧化以促进能量供应,维持心功能[8];另一方面,左心室心肌细胞PPARα 被激活后,可通过促进脂肪酸氧化减轻左心室扩张[10]。
既往实验揭示了PPARα 活性降低是代谢重塑的重要原因。sirtuin 1(Sirt1)是一种调控代谢稳态的核蛋白,是影响PPARα 活性的重要因子,可结合PPARα 靶基因启动子PPRE 区的直接重复序列1(direct repeat 1,DR1)。生理条件下,与RXRs 结合的典型DR1(typical DR1,tDR1)占优势,靶基因正常转录,从而维持脂肪酸代谢和能量供应。但随着高血压性心衰的进展,此时Sirt1 可取代RXR 结合于DR1 并抑制PPARα 靶基因转录,减弱脂肪酸氧化,使心功能恶化,见图2[11]。泛素化酶肌肉环指蛋白1(muscle RING finger protein 1,MuRF1)的上调可促进 PPARα 核输出序列 K292、K310 和 K358 三个位点的单泛素化而水解PPARα,抑制脂肪酸氧化[12]。Kruppel 样因子15(Kruppel-like factor 15,KLF15)可结合PPARα启动子,激活PPARα基因表达,改善脂肪酸代谢,延缓心衰进程[13]。
2.2 PPARγ 抑制心肌重塑 PPARγ 的激活可抑制核因子κB(nuclear factor-κB,NF-κB)p65 的核转位,减少下游通路应激相关分子表达,降低氧化应激水平,减轻心肌肥厚;PPARγ 拮抗剂可阻滞此效应,说明PPARγ 介导了对心肌重塑的抑制。而心肌细胞过表达花生四烯酸表氧化酶CYP2J2 被证明可激活PPARγ 发挥效应[14]。miR-130a 通过调控 PPARγ 的表达参与血管紧张素II的促纤维化效应[15]。
2.3 PPARβ/δ 抑制代谢重塑 有研究报道低氧条件可诱导心肌细胞miR-199a 和miR-214 表达,下调PPARβ/δ,从而促进线粒体糖酵解代谢增强,促进心衰进展。此外,上调PPARβ/δ 可促进压力负荷心衰中线粒体生成和功能,抑制代谢重塑,从而保护了心脏功能[16]。故在高血压性心衰中,激活PPARα、PPARβ/δ 和 PPARγ 可抑制心肌代谢重塑和心肌重塑,起着正向保护作用。
3 PPARs与缺血性心衰
缺血性心衰的重要机制是缺血后心肌细胞供能减少;同时氧化应激增强,促纤维化因子表达亦上调,最终引起心肌纤维化。PPARs 的表达在心梗后均有一定程度下调,而恢复PPARs水平可逆转表型,其机制如下。
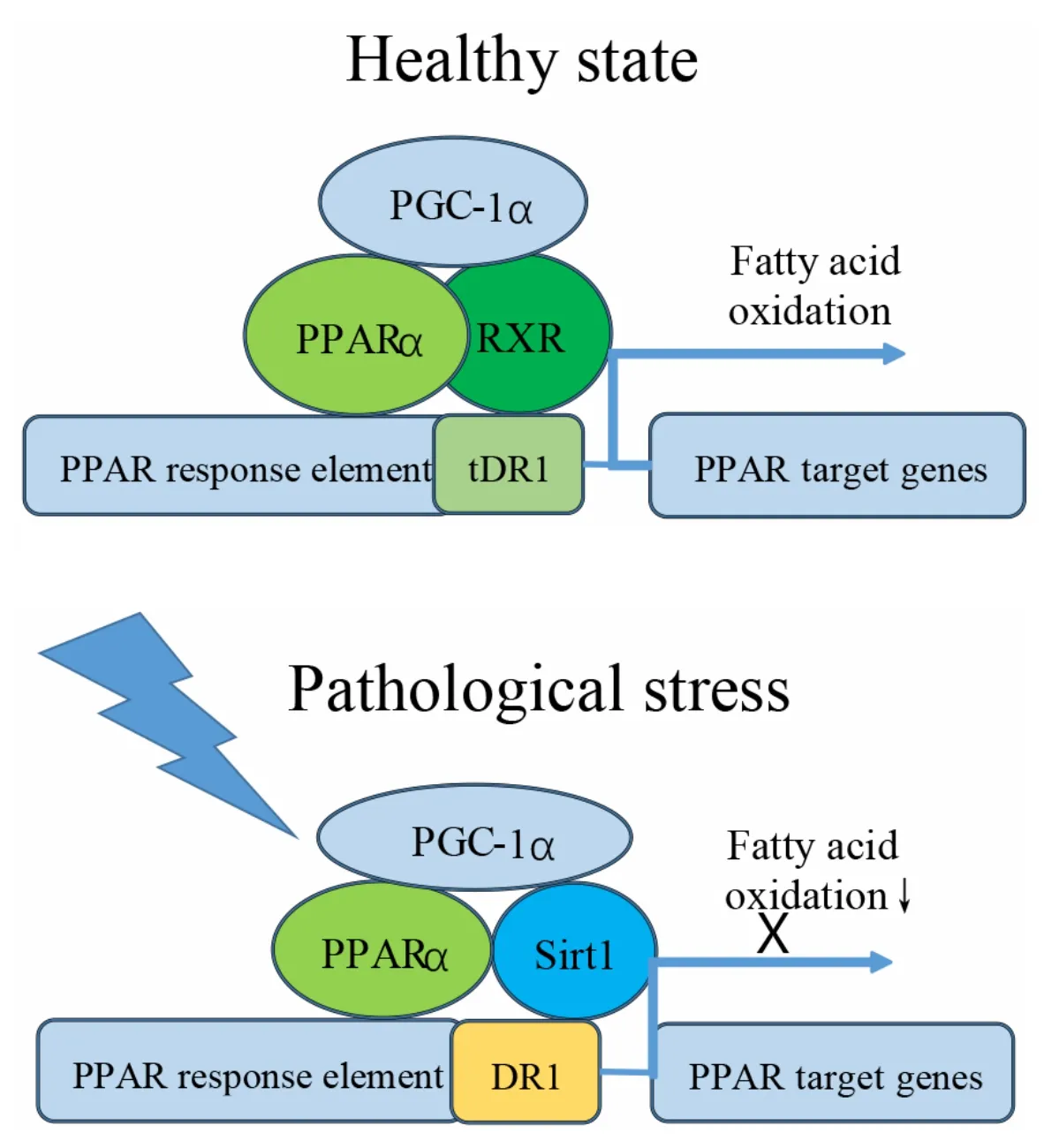
Figure 2.Sirt1 regulates PPARα in hypertensive heart failure[11].PPAR:peroxisome proliferator-activated receptor;RXR:retinoid X receptor;PGC-1α:PPARγ coactivator-1α;Sirt1:sirtuin 1;DR1:direct repeat 1;tDR1:typical direct repeat 1.图2 高血压性心衰中PPARα受到Sirt1调节
3.1 PPARα 抑制心肌纤维化 心肌纤维化的关键分子机制与NF-κB 激活有关。NF-κB 可上调下游胶原蛋白、内皮素1 等表达,促进非心肌细胞活化或增殖,分泌大量不同类型的胶原,促进心肌纤维化。PPARα 则可抑制 NF-κB 通路,减少内皮素 1 的产生,下调I/III型胶原表达,从而抑制心肌纤维化[17]。
3.2 PPARγ 改善代谢并抑制心肌纤维化 心肌梗死3 周后,中药芪苈强心可通过上调PPARγ 减缓心脏不良重塑,保留了心脏功能,减少细胞凋亡和纤维化[18]。PPARγ 抗纤维化效应亦与其抑制ERK 通路、减少成纤维细胞增殖有关[19]。
3.3 PPARβ/δ 抑制心肌氧化应激 缺血心肌常伴有抗氧化物如超氧化物歧化酶活性降低、促炎症细胞因子激活,可引起活性氧簇(reactive oxygen spe‑cies,ROS)产生增多,ROS 可损伤线粒体DNA,加重线粒体功能障碍,而PPARβ/δ 维持心肌细胞Cu/Zn/Mn 超氧化物歧化酶(superoxide dismutase,SOD)表达,减轻氧化损伤[20]。Wang 等[21]报道低氧条件可诱导心肌细胞miR-199a 和miR-214 表达,下调PPARβ/δ,减少脂肪酸氧化,促进心衰进展,而拮抗miR-199a和miR-214 可促进脂肪酸氧化而抑制代谢重塑,说明PPARβ/δ 在心衰中具有心肌保护效应。此外,卡巴环素(carbacyclin)可经PPARδ/3-磷酸肌醇依赖性蛋白激酶 1(3-phosphoinositide-dependent protein ki‑nase 1,PDK1)/第308位苏氨酸磷酸化的蛋白激酶B(phosphorylated protein kinase B at Thr308,p308Akt)/糖 原 合 酶 激 酶 3β(glycogen synthase kinase 3β,GSK3β)/β-catenin 通路促进斑马鱼和小鼠心肌细胞再生,改善心功能和远期预后[22]。
因此,激活PPARs 在缺血性心衰中可通过改善代谢、抑制心肌氧化应激、抑制心肌纤维化等机制,改善远期预后。
4 PPARs与糖尿病性心衰
糖尿病性心衰是发生于糖尿病患者且与压力负荷无关的心功能障碍,心肌脂毒性是其重要促进因素。脂毒性是指心肌细胞脂质过度积累,可增强氧化应激甚至凋亡,引起心脏功能障碍。而PPARs 在此过程中扮演重要角色。
4.1 PPARα 促进脂毒性及相关靶点 小鼠心脏特异性PPARα 过表达时,心肌细胞脂质摄取和代谢增强,糖代谢减弱,表现为糖尿病性心衰。而Nakamura等[23]发现,糖尿病时,不饱和长链脂肪酸上调心肌细胞GSK-3α,后者可特异性磷酸化PPARα配体结合结构域的Ser280,从而过度激活下游促脂质摄取途径,加剧糖尿病性心衰;非诺贝特可通竞争性抑制GSK-3α 与PPARα 的结合而逆转此效应。这些研究部分阐述了PPARα参与糖尿病性心衰发病的机制。
通过相关靶点抑制PPARα 有望成为潜在疗法:心肌细胞芳香烃受体核转位蛋白(aryl hydrocarbon receptor nuclear translocator,ARNT)/低氧诱导因子1β(hypoxia-inducible factor 1β,HIF1β)可直接结合至PPARα 起始段第二缺氧反应元件区形成稳定的复合体,抑制PPARα 转录,从而减少脂质积累[24];胰高血糖素样肽1(glucagon-like peptide-1,GLP-1)类似物exendin-4 可通过调控蛋白激酶A(protein ki‑nase A,PKA)/Rho 激酶(Rho kinase,ROCK)通路抑制PPARα介导的脂毒性[25]。
4.2 PPARγ促进脂毒性及相关靶点 Son等[26]报道了糖尿病性心衰中PPARγ 过表达,心脏特异性过表达PPARγ 亦诱发小鼠心肌脂毒性。因此,下调PPARγ 是减轻脂毒性的潜在思路,而调控PPARγ 的潜在靶点如下:GCN2(general control nondere‑pressible 2)和 1,25-二 羟 维 生 素 D3[1,25-dihy‑droxyvitamin D3,1,25(OH)2D3]均可通过下调PPARγ影响脂肪酸氧化,减轻糖尿病性心衰[27-28];MuRF2可
5 PPARs调控剂在心衰领域的转化与应用
PPARs 在心衰领域的转化前景目前在于能量代谢。PPARα 与PPARγ 在调控心肌能量代谢中存在共性和个性,与激活状态有关。PPARα 失活可减弱心肌细胞脂肪酸代谢,以致心肌细胞能量缺乏,进而加剧高血压性心衰和缺血性心衰,此类可称作“乏脂型心衰”,故上调心肌PPARα 是理论上的方案;PPARγ 亦可发挥心肌保护作用。心肌细胞PPARγ和PPARα 在特定脂肪酸下特异位点磷酸化激活,可加强心肌脂质摄取而促进心肌脂毒性,引起糖尿病性心衰,此类可视作“富脂型心衰”。而糖尿病和高血压共存时,需要更好的生物标志物或其他检查以明确心肌储脂状态而指导治疗。PPARβ/δ 仅促进脂肪酸代谢,故能量代谢方面表现为有益效应,然而治疗潜力尚待证实。而在临床上,PPARα 与PPARγ 激动剂并未应用于心衰治疗;目前还没有批准临床应用的PPARβ/δ激动剂。
贝特类是小分子PPARα 激动剂,目前用于治疗高脂血症。尽管无贝特类药物降低心衰死亡率的研究,但其安全性在荟萃分析中得到报道,其在心血管事件高风险患者一级预防中显示出良好的安全性,故非诺贝特可作为心血管疾病的一级预防[32]。
噻唑烷二酮类是PPARγ 激动剂,通过提高肝细胞胰岛素敏感性以治疗2 型糖尿病。然而其心脏作单泛素化降解PPARγ,减少脂肪酸积累而改善心功能[29]。
4.3 PPARβ/δ 与糖尿病性心衰 PPARβ/δ 具有改善脂代谢的作用。Puthanveetil等[30]报道PPARβ/δ调控脂蛋白脂肪酶(lipoprotein lipase,LPL)及其抑制剂血管生成素样蛋白4(angiopoietin-like protein 4,Angptl4),维持代谢稳态。高糖应激刺激心肌细胞释放LPL,后者可促进脂肪酸水平升高,参与糖尿病性心衰发展。而PPARβ/δ 被激活后,可解除对Angptl4的抑制,从而减少心肌细胞LPL 的转运,降低了脂代谢负荷,延缓糖尿病性心衰进程。PPARβ/δ 也有抗凋亡效应。糖尿病性心衰中凋亡因子大量激活。而PPARβ/δ 可激活过氧化氢酶基因的表达,抑制了氧化应激诱导的线粒体源性凋亡;亦可减轻内质网应激,上调自噬相关分子,从而减少了慢性炎症诱发的细胞凋亡[31]。
因此,在糖尿病性心衰中,激活PPARγ 和PPARα 可促进脂毒性而加重心衰,激动PPARβ/δ 则通过促代谢和抗凋亡而维持心功能。
表1 总结了PPARs 在心衰中研究进展的分子机制。用仍存在重大争议。既往已观察到与罗格列酮给药有关的心脏事件增多,可能与其促进心肌神经酰胺和脂肪酸积累引起的脂毒性有关,2010 年欧洲药品管理局因此撤销了罗格列酮的批准。因此,有必要进一步研究PPARγ激动剂的安全性。
总之,PPAR 调控剂在心力衰竭中的临床应用尚未完全起步,仅部分PPARα 激动剂投入市场,能否作为安全有效的靶点指导临床治疗尚待验证。
6 总结与展望
在高血压性心衰和缺血性心衰中,PPARs 均发挥正向保护作用,可通过促进脂肪酸氧化促进代谢供能,抑制氧化应激并纤维化,抗凋亡及减轻心肌肥厚。而在糖尿病性心衰中,PPARβ/δ 仍可通过上述机制改善心功能,PPARα和PPARγ的过表达或激活在动物模型和心肌细胞中却表现为负向损害作用,促进脂毒性而加重心衰,然而前两种心衰中PPARα降低的机制,以及糖尿病性心衰中PPARα 和PPARγ对心肌作用的详细机制有待进一步研究。
临床上PPARα 和PPARγ 激动剂并未在多种心衰治疗中展现疗效,甚至带来可能产生心衰的副作用。故只有针对不同类型心衰、不同机制合理调控PPARs,才可能使其成为未来治疗心衰的有效潜在靶点。

表1 PPARs相关新机制Table 1.Novel mechanisms associated with PPARs.
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国慈善家(2021年5期)2021-11-19
世界科学技术-中医药现代化(2021年7期)2021-11-04
食品安全导刊(2021年20期)2021-08-30
中华养生保健(2020年9期)2021-01-18
科学咨询(2020年10期)2020-04-01
科学咨询(2020年1期)2020-02-11
自我保健(2019年10期)2019-12-11
英语文摘(2019年6期)2019-09-18
中国实验动物学报(2019年4期)2019-09-03
