
遗传性球形红细胞增多症
——一家系中SPTA1基因突变分析及产前诊断
2020-09-04 08:52叶玉萍马诗玥廖林黄健邓雪莲林发全
广西大学学报(自然科学版) 2020年4期
叶玉萍,马诗玥,廖林,黄健,邓雪莲,林发全*
(1.广西医科大学第一附属医院检验科,广西南宁530021;2.桂林医学院附属医院检验科,广西桂林541000)
遗传性球形红细胞增多症(hereditary spherocytosis, HS)是一种红细胞膜蛋白结构异常所致的遗传性溶血病,其主要临床表现为贫血、黄疸、脾肿大和胆石症,血液学特征为外周血中出现较多小球形红细胞。 HS的基本发病机制是由于编码红细胞膜蛋白的基因突变导致红细胞膜结构与功能异常,其主要致病基因包括SPTA1基因、SPTB基因、SLC4A1基因、ANK1基因、EPB42基因,分别编码α-血影蛋白、β-血影蛋白、带3蛋白、锚蛋白和4.2蛋白5种红细胞膜蛋白[1]。α-血影蛋白是一种细胞骨架蛋白,其与β-血影蛋白结合形成反平行的α-β异二聚体,两分子α-β异二聚体头对头连接形成四聚体,通过与红细胞内肌动蛋白、带3蛋白、锚蛋白、4.2蛋白等相互作用,维持红细胞膜结构稳定性和变形能力[2]。
编码α-血影蛋白的SPTA1基因位于染色体1q22-q23区,包含52个外显子。SPTA1基因突变所致的HS约占遗传性球形红细胞增多症病例的5 %,且多数患者仅在纯合或复合杂合突变时才表现出明显的临床症状。 近年来,国内外有关SPTA1基因突变引起HS的研究报道较少。本研究对1例HS患者及其父母的SLC4A1、ANK1、SPTA1、SPTB、EPB42基因各外显子的编码区及剪接区的序列变异情况进行突变分析,并对患儿母亲的胎儿进行产前基因检测,以探讨HS分子遗传学特征和产前诊断的临床意义[3-5]。
1 资料与方法
1.1 一般资料
患儿(图1 Ⅱ1),女,6岁,贵州籍,因“反复皮肤黄染6年伴乏力”入院。出生后48 h出现贫血伴黄疸,曾予输血治疗,效果不佳。
入院查体:一般状态尚好,心肺未见异常,全身皮肤巩膜轻度黄染,腹软,触诊肝大,肋下约2.0 cm;脾大,肋下约4.0 cm,腹部B超提示“脾肿大”,肝、胆、胰回声未见明显异常,个人史家族史无特殊。实验室检查:红细胞、血红蛋白、红细胞平均体积、红细胞平均血红蛋白含量结果分别为1.85×1012/L、48.10 g/L、77.26 fL、26.06 pg;总胆红素、直接胆红素、间接胆红素结果分别为96.83 μmol/L、14.94 μmol/L、81.89 μmol/L。尿胆红素(3+),G-6-PD酶活性正常,G-6-PD和地中海贫血基因分析未见异常,外周血涂片检查结果显示球形红细胞达45 %。结合临床表现和实验室检查结果诊断为HS。完善相关检查后,行腹腔镜脾脏切除术,术中病理结果符合HS引起的脾肿大。术后1个月复查,患儿血红蛋白浓度升至131.30 g/L,外周血球形红细胞比例降至25 %(图2)。4年后,患儿母亲再次妊娠,转诊到我院进行HS产前遗传咨询。患儿监护人及家系成员均签署知情同意书。

图1 HS患者家系系谱图Fig.1 Pedigree chart of HS pedigree
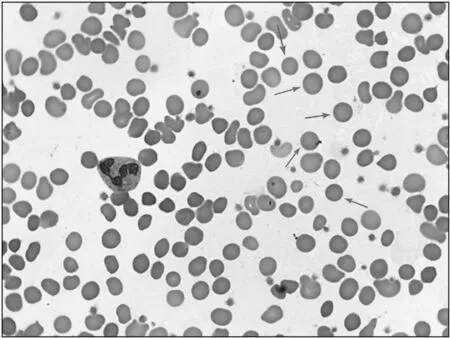
箭头所示为典型的球形红细胞
1.2 方法
1.2.1 α-血影蛋白分析
应用蛋白免疫印迹法(Western blot)检测患儿及其父母红细胞膜蛋白中α-血影蛋白表达水平。参考文献[6]提出的方法配制SDS-PAGE分离电泳凝胶。80 V 转 120 V 电泳4 h,转膜2 h,5 % 脱脂奶粉封闭1 h,4 ℃孵育一抗过夜,第二天回收一抗,TBST洗膜3次,室温孵育二抗 1 h,TBST 洗膜后在Fluor ChemM多色荧光和化学发光成像系统显影。
1.2.2 基因检测分析
采集患儿及其父母外周静脉血2 mL,EDTA抗凝,采用DNA提取试剂盒(北京天根生化科技公司)提取外周血基因组DNA。通过高通量测序检测患儿外周血样本提取的基因组DNA中SLC4A1,ANK1,SPTA1,SPTB和EPB42基因各外显子的编码区及剪接区的序列变异情况。患儿母亲妊娠21周,在B超引导下行羊膜腔穿刺术,抽取羊水10 mL,离心后分离细胞,用上述试剂盒提取羊水细胞DNA,应用PCR结合Sanger测序确认突变位点。
1.2.3 突变致病性预测
使用基因突变致病性预测软件PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml)和Mutation Taster (http://www.mutationtaster.org/)对突变位点的致病性进行预测。 如果两个软件预测结果均显示为“有害”,则认为该突变可能具有致病性。
2 结果
2.1 临床随访实验室检查结果
术后4年,患儿母亲再次妊娠,转诊到我院进行HS产前遗传咨询。患儿及其父母的实验室检查结果显示,患儿总胆红素和直接胆红素水平超过正常范围两倍,其父母的胆红素水平正常(表1)。
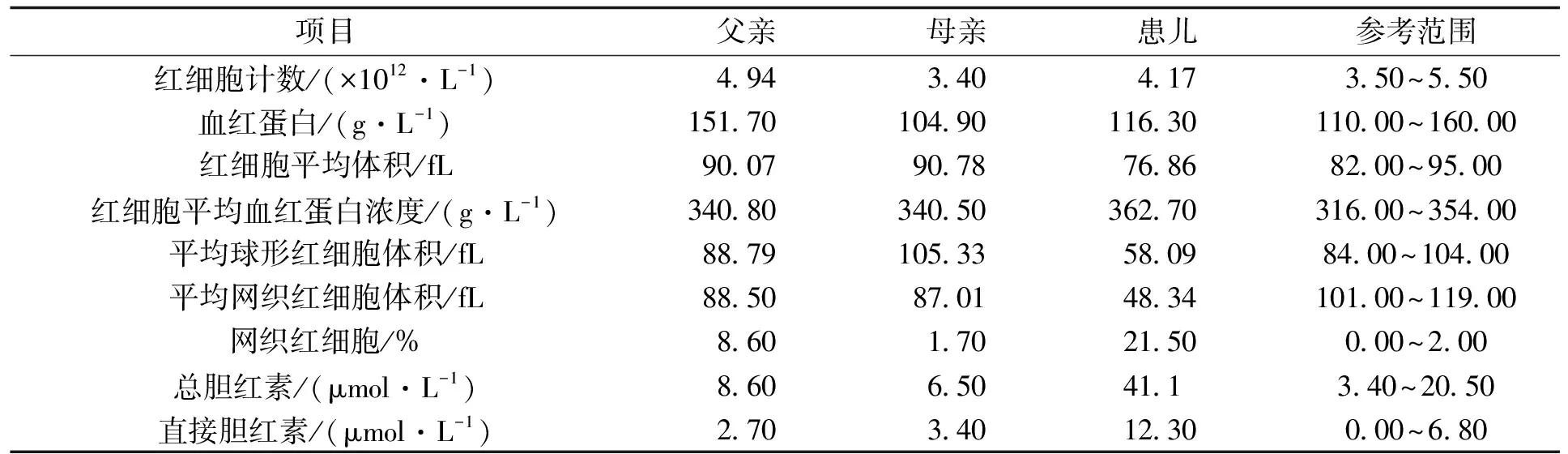
表1 脾切除术后4年患儿及其父母的实验室检查结果Tab.1 Laboratory test results of the proband and her family in the fourth year after splenectomy
2.2 α-血影蛋白分析结果
Western blot结果表明,患儿母亲α-血影蛋白表达量与正常人相比未见明显差异,而HS患儿及其父亲的α-血影蛋白表达量下降(图3)。
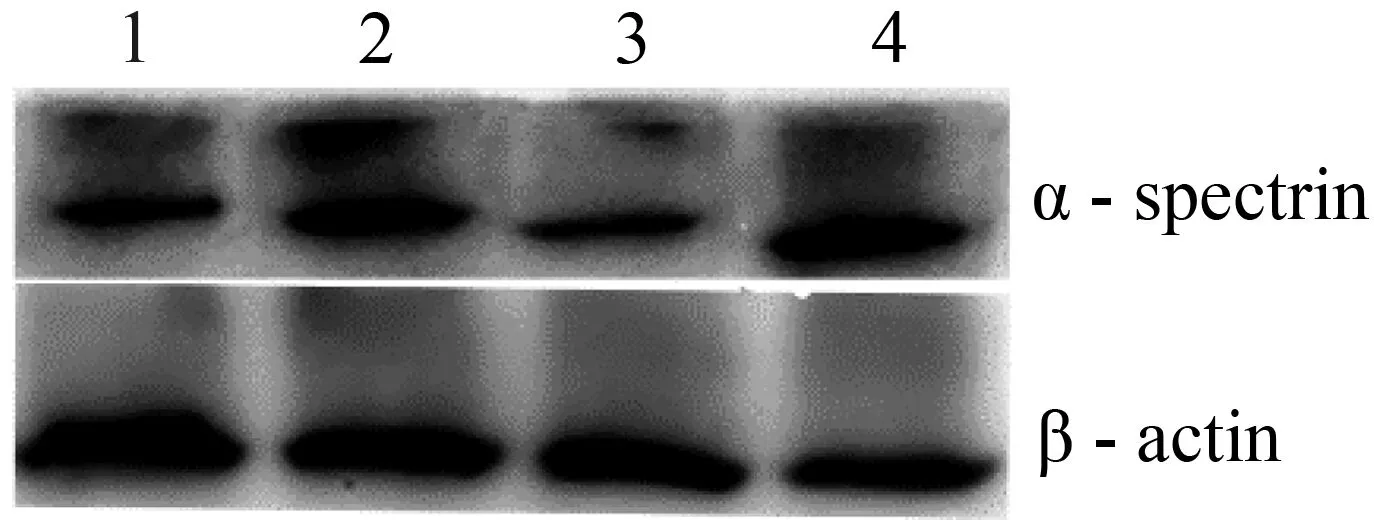
1~4分别为患儿父亲、患儿母亲、患儿和正常对照
2.3 基因检测结果
高通量测序结果显示患儿HS相关的5个基因中,SPTA1基因检测到第16外显子c.2179_2180delAG(p.Arg727Glufs*27)移码突变和第26外显子c.3710A>G(p.Asp1237Gly)错义突变。PCR结合Sanger测序法证实了患儿SPTA1基因中这两个突变位点的存在,并根据已知位点对其父母SPTA1基因进行检测,发现其父母分别为c.2179_2180delAG(p.Arg727Glufs*27)移码突变和c.3710A>G(p.Asp1237Gly)错义突变的杂合子携带者[图4(a)~(d)],该HS家系SPTA1基因测序分析结果示于表2。第16外显子c.2179_2180delAG(p.Arg727Glufs*27)移码突变,导致精氨酸突变为谷氨酸,造成氨基酸编码提前终止[图4(a)~(b)]。第26外显子c.3710A>G(p.Asp1237Gly)错义突变,导致α-血影蛋白中第1237位的天冬氨酸被甘氨酸取代[图4(c)~(d)]。上述两个突变均为新突变,在人类基因突变数据库(HGMD; http://www.hgmd.cf.ac.uk/ac/index.php)等相关数据库及医学文献中未见报道。
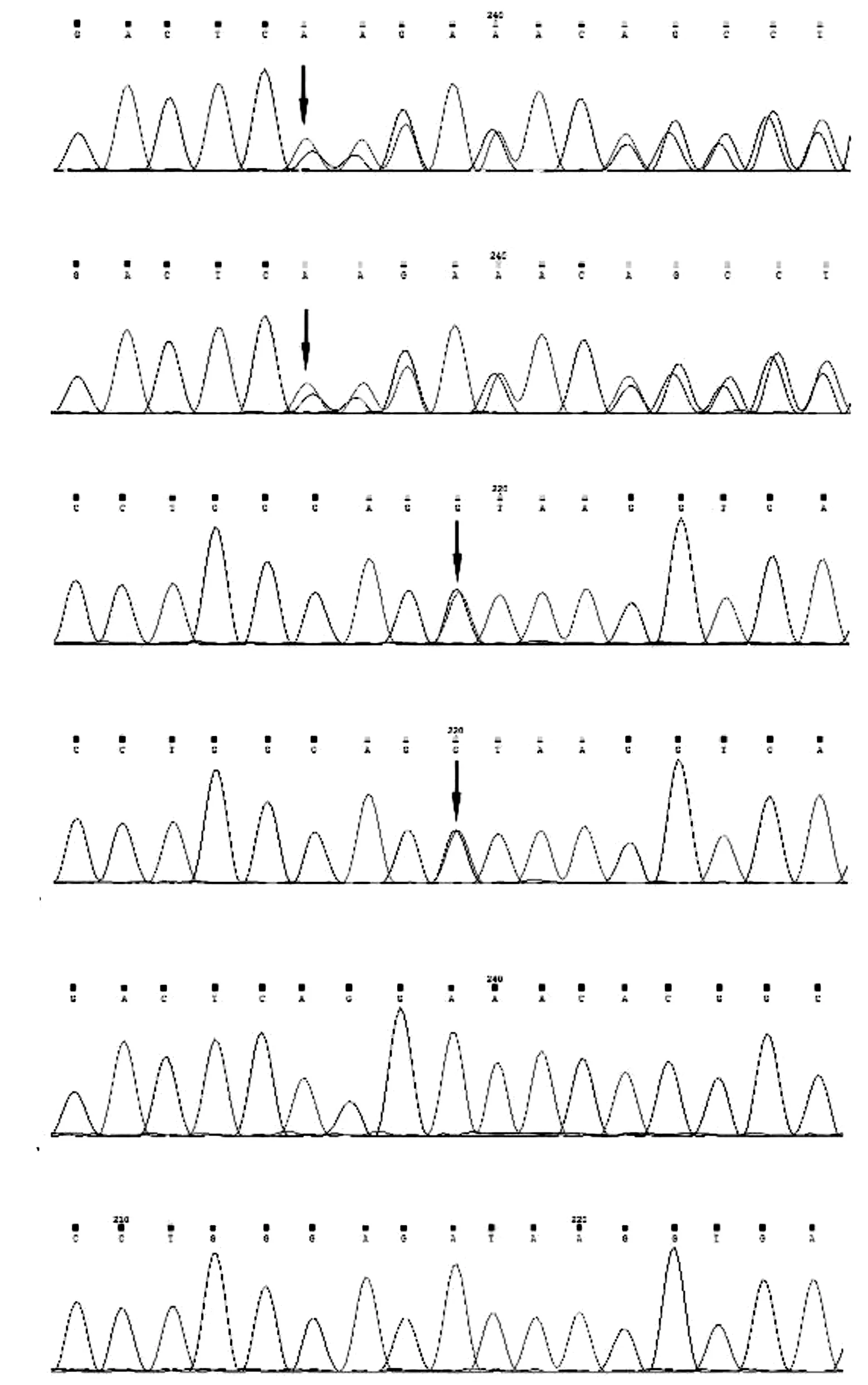
(a) 箭头所示为突变位点:患儿父亲第16外显子c.2179_2180delAG移码突变 (b) 患儿第16外显子c.2179_2180delAG移码突变 (c) 患儿母亲第26外显子c.3710A>G 错义突变 (d) 患儿第26外显子c.3710A>G 错义突变 (e)胎儿羊水细胞SPTA1基因第16外显子基因测序结果未检测到来自父母的SPTA1基因突变 (f) 胎儿羊水细胞SPTA1基因第26外显子基因测序结果未检测到来自父母的SPTA1基因突变

表2 该HS家系中检测出的SPTA1基因突变Tab.2 SPTA1 gene mutations identified in this pedigree
2.4 突变致病性预测结果
运用PolyPhen-2和Mutation Taster对SPTA1基因中两个新突变的致病性进行预测,PolyPhen-2软件预测c.3710A>G错义突变为致病性突变的可能性小,而Mutation Taster软件预测结果则认为该突变可能为致病性突变。 c.2179_2180delAG为移码突变,可能会造成氨基酸编码提前终止,产生截短蛋白。PolyPhen-2和Mutation Taster预测结果均表明c.2179_2180delAG为致病性突变的可能性大。
2.5 产前诊断结果
患儿母亲羊水细胞DNA基因分析未检测到来自父母的SPTA1基因突变[图4(e)~(f)],胎儿为SPTA1基因c.2179_2180delAG(p.Arg727Glu fs*27)杂合变异导致的HS患者的可能性小。 经遗传咨询,患儿母亲继续妊娠并分娩了健康女婴,其与HS相关的实验室检查结果均在正常范围内:血红蛋白122.00 g/L,红细胞计数5.19×1012/L,总胆红素19.41 μmol/L,直接胆红素5.69 μmol/L。
3 讨论
HS的分子发病机制是基因突变导致红细胞膜蛋白缺陷,与红细胞膜脂质双分子层垂直联接作用减弱,引起红细胞膜脂质双分子层稳定性降低,膜脂质以微囊泡的形式释放,使红细胞表面积减少,红细胞呈球形改变[7-8]。HS患者红细胞膜结构完整性受损与膜脂质结合的丙酮酸激酶(PK)丢失而引起PK活性降低有关,可能会加剧溶血的严重程度[9]。HS的遗传方式多为常染色体显性遗传,约占HS患者的75 %,另约 25 % 的患者无家族史,可表现为自发突变或隐性遗传。α-血影蛋白缺陷引发的HS为常染色体隐性遗传,而β-血影蛋白缺陷引发的HS为常染色体显性遗传,因为α-血影蛋白的表达量为β-血影蛋白的4倍,故α-β异二聚体的形成不受α-血影蛋白表达量相对降低的影响。根据患者血红蛋白、胆红素浓度及网织红细胞计数,HS可以分为轻度,中度,中重度和重度,许多轻度HS患者临床症状不明显且缺乏特异性,易被忽视。中重度HS患儿可能表现出精神运动发育迟缓[10-11]。SEREGINA等[12]研究发现HS患者并发血栓的风险增加,而血栓弹力图(TEG)和血栓动力学分析(TD)有利于监测HS患者的凝血系统以预测血栓形成倾向。
HS在世界范围内均有报道,在北欧人群中,HS是最常见的先天性溶血性贫血,发病率为1/2 000[13],同时HS也是日本最常见的先天性溶血性贫血[14]。通过DisMod-II软件评估中国人1978~2013年HS的患病率,男性为1.27/100 000;女性为1.49/100 000[15]。 然而,中国HS患者中SPTA1基因突变的报道较少。WANG等[16]报道了国内1例Coombs实验阴性的HS患者,发现SPTA1基因中2个新突变(c.3897-1G> C和c.5029G> A)。SPTA1基因第27-1内含子中的c.3897-1G> C突变破坏了共有的剪接位点,为主要致病突变。而c.5029G> A(p.Gly1677Arg)突变则导致α-血影蛋白第1677位的甘氨酸被精氨酸取代。PolyPhen-2,SIFT和MutationTaster软件预测结果均表明,c.3897-1G> C和c.5029G> A可能为致病性突变。
脾切除术适用于中度至重度HS患儿,但过早切脾会使机体免疫力下降,易引发严重感染,因此原则上应在6岁后行脾切除术,以降低脾切除术后败血症的发生率[17]。本研究中,术后第4年复查,患儿的高胆红素血症已得到一定程度的纠正,但网织红细胞,总胆红素和直接胆红素水平仍高于正常范围,这与MARIANI等[18]先前报道的血影蛋白缺陷HS患者脾切除后的情况相符。
本研究发现SPTA1基因中的2个新突变,分别为c.2179_2180delAG(p.Arg727Glufs*27)移码突变和c.3710A>G(p.Asp1237Gly)错义突变,c.2179_2180delAG(p.Arg727Glufs*27)移码突变导致突变位点后第27个密码子存在终止密码子,SPTA1基因翻译提前终止,产生截短蛋白,影响α-血影蛋白的功能。PCR结合Sanger测序法证实了患儿SPTA1基因中这2个突变位点分别来自其父母,并对再次妊娠的患儿母亲羊水细胞进行基因分析,对胎儿成功进行了产前诊断。由于HS患者的临床表现具有明显的异质性,易造成漏诊和误诊,因此基因检测对HS的确诊起重要作用。此外,HS的热点突变较少,绝大多数报道的突变为HS家系所特有的新突变。对HS家系相关致病基因进行突变检测,可发现无症状或症状轻微的患者及致病基因携带者,为遗传咨询和产前诊断提供依据。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
中成药(2017年12期)2018-01-19
中国生殖健康(2018年2期)2018-01-12
川北医学院学报(2015年5期)2015-12-05
西藏科技(2015年5期)2015-09-26
湖北农业科学(2014年11期)2014-09-10
