
ICP-MS法测定1β-甲基碳靑霉烯双环母核原料药残留
2020-09-03 08:28池海涛李琴梅刘奕忍
分析仪器 2020年4期
池海涛 赵 婷 李琴梅 刘奕忍 高 峡 徐 聪
(北京市理化分析测试中心,有机材料检测技术与质量评价北京市重点实验室,北京 100094)
原料药1β-甲基碳靑霉烯双环母核(MAP),中文名称美罗培南主环,CAS:90776-59-3,分子式:C29H27N2O10P,相对分子量:594.14,是日本住友公司首先研发的新型碳靑霉烯抗生素,具有广谱,强效,耐药性好等优点[1,2]。在其合成过程中,需要加入一定量的贵金属催化剂铑,因此在其合成好的原料药中会有催化剂铑的残留。催化剂铑的残留含量测定是MAP原料药质量及药物安全性评价的一个重要指标。目前药品中催化剂残留的测试方法主要是利用原子光谱类仪器进行微量分析,有AAS(石墨炉原子吸收光谱)、ICP-OES(电感耦合等离子体发射光谱)、ICP-MS(电感耦合等离子体质谱)法等[3-13]。其中AAS法数据稳定性差,需要基体改进剂,费时费力。ICP-OES法检出限只能达mg/kg级别,无法满足药物中痕量元素测定。ICP-MS法检出限可以达到μg/kg,具有稳定性好,简便快速等优点,可以药物元素催化剂残留的要求。由于药物的原料药有机物结构式每种都不同,因此催化剂残留检测方法需要针对不同的药物结构式进行开发,本实验中研究的原料药中铑催化剂的残留含量分析方法文献中涉及的较少,分析方法需要进一步针对性的优化。本实验采用ICP-MS法研究开发MAP(1-β甲基碳靑霉烯双环母核)原料药中催化剂残留铑的含量,通过优化前处理方法,寻找简便的消解方法,同时通过ICP-MS分析方法的开发来找到更优的方案来进行MAP(1-β甲基碳靑霉烯双环母核)原料药中催化剂残留铑的含量的测试分析。
1 实验部分
1.1 仪器与试剂
7700x电感耦合等离子体质谱仪:美国Agilent公司,配有自动进样器及MassHunter软件操作系统;Mettler Toledo XS204电子分析天平:瑞士梅特勒-托利多公司,最小分度0.1mg;Milli-Q超纯水处理系统:美国Millipore 公司; PXS-215型离子酸度计:上海雷磁;99.99%高纯液氩:北京永圣气体科技有限公司,DigiBlock EHD36电热消解仪(莱伯泰科公司)
硝酸为Mos级,购自于美国Merck公司;高氯酸为优级纯,购自北京化学试剂厂;锂、钴、铊、铈、铱调谐溶液(10μg/L),均购自美国Agilent公司;实验过程中所用超纯水电阻率为18.2 MΩ·cm,消解管、容量瓶等均用HNO3(1+4)浸泡24 h以上用超纯水冲洗后备用。1β-甲基碳靑霉烯双环母核(MAP)原料药为某药厂提供;铑标准样品(103Rh):1000(mg/L),购自国家钢铁材料测试中心;优级纯硝酸购自MERCK公司;高氯酸购自国药集团。
1.2 样品前处理
采用湿法消解(敞口)进行样品消解,称取0.20 g样品于石英消解管中,先加入5mL Merck级硝酸于150℃进行湿法消解,待消解至1mL以下时(约3小时),加入少量二次纯水,有沉淀生成证明其消解不完全;故再加入5mL硝酸消解至近干(即1.0mL以下);此时再加入2.0mL优级纯高氯酸继续消解直至澄清透明(约为4小时)。待消解完全并赶酸至近干后冷却,用去离子水转移定容至25mL容量瓶中,定容、摇匀;采用0.22μm的医用注射针头滤膜进行过滤后,等待上机。
1.3 仪器操作条件
仪器操作:采样锥及截取锥的长期使用,表面和锥孔孔径会发生一定的变化,因此测试前需采用1.0 μg/L的锂、钴、铊、铈调谐液对仪器进行自动调谐,通过调整矩管位置,离子透镜电压,质量轴等,使仪器的灵敏度、分辨率、氧化物及双电荷产率等各项指标达到测定要求。调谐优化后的ICP-MS参数如表1所示。该仪器状态满足测试要求。

表1 ICP-MS的工作参数
2 实验结果与讨论
2.1 标准曲线的确定
采取重量法配制一系列标准溶液,分别量取一定体积浓度为10mg/L 铑标液(Agilent,#8500-6948),记录质量;用去离子水稀释至相同质量后计算相应浓度,所得浓度分别为10.0646、21.433540.9489、59.8356、80.6336μg/L。得出标准工作曲线及线性关系(采用In在线内标)如下:y=0.3740x+ 0.0025,R2=0.9997;从而可知该方法线性良好。
2.2 基体干扰的消除
ICP-MS虽然具有很好的抗干扰性,但是在分析过程仍然受到质谱干扰和非质谱干扰的影响,样品的基体效应、同质异位素、多原子离子、氧化物等都是影响ICP-MS结果准确性的关键因素[11]。MAP样品ICP-MS分析过程中的潜在的干扰主要是基体效应造成,干扰易造成仪器信号漂移,且干扰程度与基体性质相关。一般采用在线内标校正措施来降低乃至消除干扰。为了避免样品基体对测试的干扰,一般可以采用Li、Sc、Ge、Rh、In、Bi、Tb作为内标的内标法对元素进行测定,本实验中对于铑元素的测定则以In做在线内标法。
2.3 专属性实验
连续进11次空白溶液,测定结果如表2所示,其中响应值偏差(δ)为 0.00154,表明该方法专属性良好。
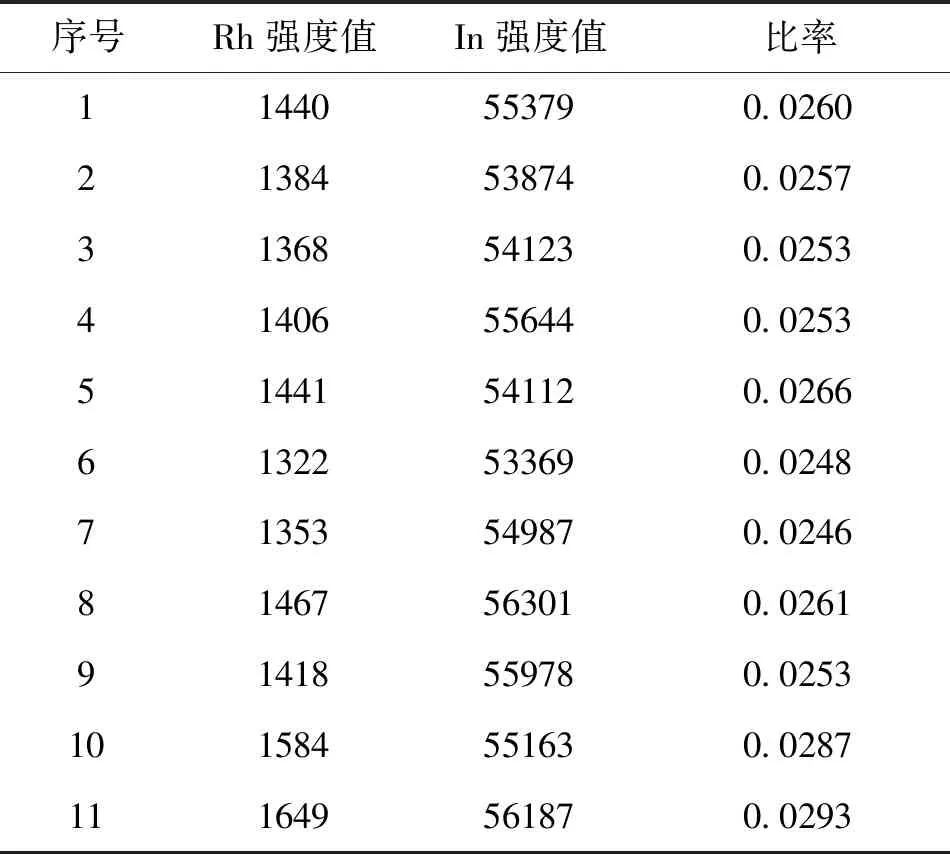
表2 专属性结果表
2.4 精密度实验
以MAP作为样品进行方法学研究,开展样品精密度实验。分别做仪器和方法的精密度实验。
2.4.1仪器的精密度实验
取(限度浓度约10.0 mg/kg)溶液浓度约为80.0μg/L的铑标准溶液,连续测定20次强度值,计算出RSD为3.76%,说明仪器精密度良好(表3)。
表3 精密度实验
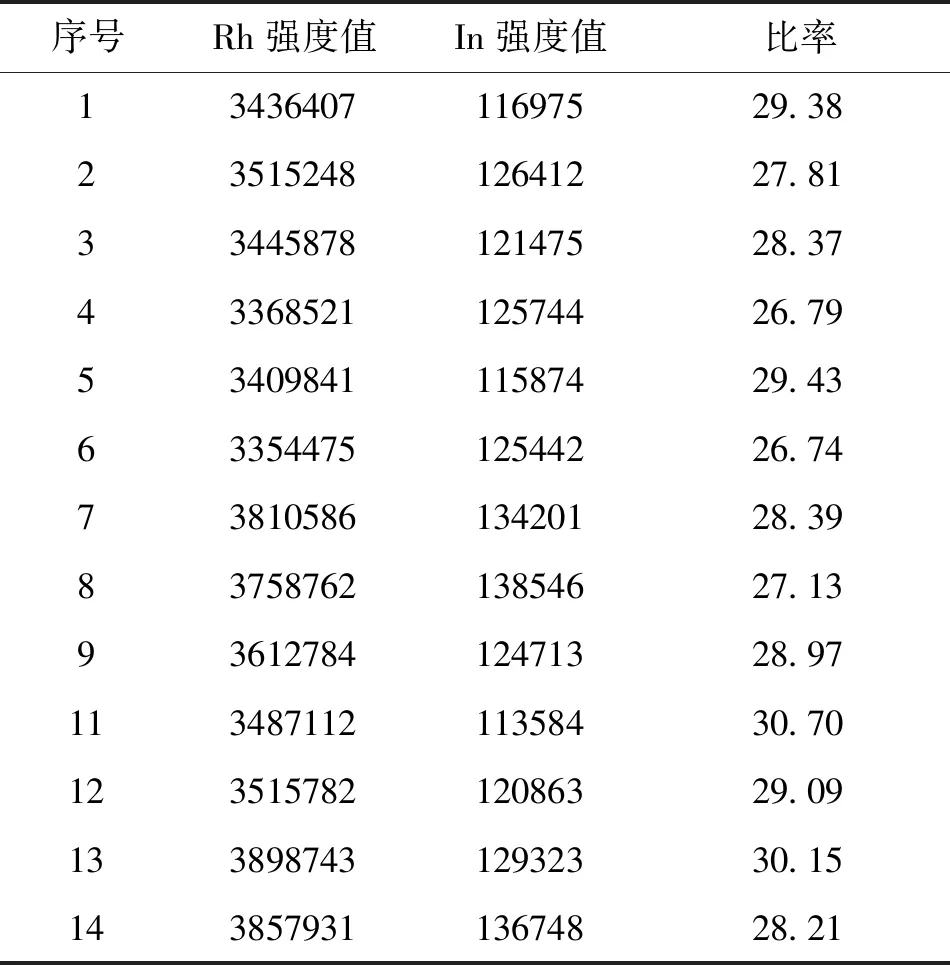
序号Rh强度值In强度值比率1343640711697529.382351524812641227.813344587812147528.374336852112574426.795340984111587429.436335447512544226.747381058613420128.398375876213854627.139361278412471328.9711348711211358430.7012351578212086329.0913389874312932330.1514385793113674828.21

续表3
2.4.2方法的精密度实验
称取6个样品每个0.20 g左右于消解管中,采用硝酸加热消解样品,赶酸至样品澄清透明,定容至25mL,过滤,同时做样品空白。ICP-MS测定结果如表4所示。分析结果的精密度用相对标准偏差来衡量,由表4可知,浓度含量计算出RSD为0.95%,说明分析方法的精密度较好。

表4 精密度实验结果
2.5 检出限
按照中华人民共和国药典2015年版4部《药品质量标准分析方法验证指导原则》之规定如下:检出限计算公式:LOD=3.3*δ/S;定量限计算公式:LOD=10*δ/S。
由2.3专属性实验得出其中响应值比率标准偏差(δ):0.00154;由2.1得出标准曲线斜率(S):0.3740。
计算检出限LOD为0.0136μg/L,计算定量限LOQ为0.0412μg/L。
参考其他文献中最低检出质量浓度计算方法[12],其中最低检出质量浓度以称样0.2g消解定容至25mL计,得出最低检出质量浓度为:LOD*25mL/0.2g=1.70μg/kg。
2.6 回收率实验
以MAP作为样品进行方法学研究,开展样品回收率实验。
称取样品0.20 g于消解管中,分别加入0.200、0.300、0.400μg铑(并分别做3个平行),采用硝酸加热消解样品,赶酸至样品澄清透明,定容至25mL,过滤,对其进行加标回收率实验。加标回收实验结果如表5所示,回收率都在90%以上,表明分析方法可行。

表5 回收率实验结果
2.7 样品测定与实验结果
通过上述对MAP样品进行平行性和回收率实验,结果表明:该测试方法可以用于原料药 1β-甲基碳靑霉烯双环母核(MAP)中催化剂铑残留的测定。具体结果如表6所示。

表6 样品测试结果
3 结论
原料药 1β-甲基碳靑霉烯双环母核中催化剂铑残留是药物质量控制中一个重要指标,本实验建立了ICP-MS测定1β-甲基碳靑霉烯双环母核中铑残留的方法。通过配制铑系列标准溶液,验证标准曲线方程和相关系数,确定该方法的相关系数均为0.999以上。利用该检测方法,铑回收率较好,在90.88%~100.40%之间。方法检出限可达到1.70μg/kg,精密度实验RSD在4%以内。本实验提出的ICP-MS测试法快速、灵敏、准确,可以完全用于原料药1β-甲基碳靑霉烯双环母核中催化剂铑残留的测定,可为药品的质量控制提供方法依据。
猜你喜欢
安徽农学通报(2022年8期)2022-05-06
检验医学与临床(2020年1期)2020-01-10
中国盐业(2018年20期)2019-01-14
家用汽车(2016年12期)2017-02-09
应用海洋学学报(2015年2期)2015-11-22
中国当代医药(2015年33期)2015-03-01
现代检验医学杂志(2015年6期)2015-02-06
中国医疗美容(2015年5期)2015-02-03
中华皮肤科杂志(2014年4期)2014-12-19
