
铈掺杂对介孔硅锆复合氧化物酸性的影响
2020-08-21 14:56:02唐芳玲郭建忠王志坤
石油化工 2020年7期
李 莎,唐芳玲,郭建忠,王志坤
(浙江农林大学 化学系,浙江 杭州 311300)
酸催化在当代化学工业中占有重要地位。SiO2-ZrO2固体酸因为具有热稳定性高、机械强度高、热扩散性低、污染小、易回收等优点而备受关注。硅锆比[1-2]、材料晶型[3]、焙烧温度和气氛[4]等是影响SiO2-ZrO2酸性的主要因素,一些后处理方法也会影响SiO2-ZrO2的酸性:甲基化会明显降低材料的质子酸位点[5]、嫁接硫酸盐能提高材料的酸性[6]、添加Cu[7]和Mo[8]等有助于形成强酸位点。
在分子筛中加入Ce元素会提高分子筛的酸催化活性,稀土元素经过焙烧会迁移到更小的笼表面与氧形成桥键,再发生水解,生成质子酸位点[9]。NH3-TPD表征结果证明,ZSM-5和SAPO-34分子筛负载2%(w)Ce3+后,材料的中强酸和强酸含量均变多[10],但将Ce掺杂到ZSM-5分子筛中会降低材料的弱酸强度及L酸含量[11]。将Ce加入到HBEA分子筛中会减少它的B酸位点[12]。关于Ce掺杂对介孔SiO2-ZrO2酸性的影响研究较少。
本工作采用溶胶-凝胶法合成了介孔SiO2-ZrO2材料,对其进行了N2吸附-脱附、XRD、吡啶吸附FTIR(Py-FTIR)和XPS表征,并将其用于苯甲醚和苯甲醇的傅克烷基化反应,研究了Ce掺杂和焙烧温度对SiO2-ZrO2酸性的影响。
1 实验部分
1.1 主要试剂
盐 酸、冰 醋 酸、乙 醇、正 硅 酸 乙 酯、CeCl3·7H2O:分析纯,国药集团化学试剂有限公司;正丁醇锆:80%(w)正丁醇溶液,阿拉丁试剂有限公司;三嵌段共聚物F127(EO106PO70EO106,EO为环氧乙烷,PO为环氧丙烷):分析纯,摩尔质量为12 600 g/mol,Sigma公司。
1.2 催化剂的制备
按照文献[13]报道的方法合成介孔SiO2-ZrO2(Zr/Si摩尔比为1)。25 ℃下,在30 mL无水乙醇中加入1.6 g表面活性剂F127,再加入1 mL浓盐酸、2.3 mL冰醋酸,搅拌;待溶液澄清后加入1.114 mL正硅酸乙酯、2.286 mL正丁醇锆、0.039 7 g的CeCl3·7H2O,继续搅拌2 h后得到溶胶体系;将溶胶倒入培养皿中自然挥发直至成膜状,然后在65 ℃下老化24 h,再在马弗炉中焙烧5 h,得到Ce掺杂的SiO2-ZrO2催化剂,记为ZS-Ce-T,T表示焙烧温度,℃。ZS-Ce-T催化剂中Ce的质量分数为2%。
1.3 催化剂的表征
采用Quantachrome公司NOVA 4200e型自动吸附仪进行N2吸附-脱附表征。试样预先在200 ℃、真空条件下预处理4 h,通过BJH模型和吸附曲线计算孔径分布和孔体积。
采用Bruker公司D8 Advance型X射线粉末衍射仪进行XRD表征。CuKα射线,波长0.154 18 nm,管电压为40 kV,管电流为30 mA,扫描步长为0.02°,扫描速率为4 (°)/ min。
采用Bruker公司TENSOR 27型傅里叶变换红外光谱仪进行Py-FTIR表征。试样在300 ℃、真空条件下保持1 h,预处理后降至室温;升温至150 ℃后通入吡啶,吸附30 min,然后在150 ℃下抽真空1 h,以去除物理吸附的吡啶,降至室温后开始测试。
采用Thermo Scientific公司ESCALAB 250Xi型X射线光电子能谱仪进行XPS表征。AlKα射线(hν=1 486.6 eV),以C1s284.8 eV为基准进行结合能校正,采用XPS Peak4.1软件对数据进行分峰拟合。
1.4 酸催化反应
在50 mL圆底三颈烧瓶内加入0.1 g催化剂、50 mmol(5.445 mL)苯 甲 醚 和5 mmol(0.518 mL)苯甲醇,搅拌,置于恒温油浴中,在154 ℃下开始反应,每隔一定反应时间取20 μL溶液溶解于800 μL无水乙醇中,进行气相色谱分析。气相色谱分析条件:上海奇阳信息科技有限公司GC-9860型气相色谱仪,SE-54石英毛细管柱(30 m×0.32 mm×0.50 μm),FID检测;柱温采用程序升温方式,从80 ℃到250 ℃,升温速率为20℃/min;进样器和检测器温度均为250 ℃;以正癸烷为内标物,采用校正面积归一化法。
通过苯甲醇的转化率来评价催化剂的性能,苯甲醇的转化率(X)和产物的选择性(S)计算如下:

式中,nI表示反应了的苯甲醇的物质的量,mol;nF表示加入的苯甲醇的物质的量,mol;nP表示生成的邻苄基苯甲醚和对苄基苯甲醚的物质的量,mol。
2 结果与讨论
2.1 Ce掺杂的影响
SiO2-ZrO2材料常用的合成方法有共沉淀法、机械混合法、浸渍法和溶胶-凝胶法[14]。相比于其他方法,溶胶-凝胶法制备的催化剂具有均匀性更好、比表面积更大、孔径可调节等优势[15]。本工作选用溶胶-凝胶法合成SiO2-ZrO2介孔材料。为了对比,制备了不掺杂Ce的SiO2-ZrO2催化剂,记为ZS-T。
苯甲醇和苯甲醚的傅克烷基化反应需要在中强B酸或强B酸位点上进行[16],且随着酸量或酸强度的增加,催化活性提高,反应产物为邻苄基苯甲醚和对苄基苯甲醚,主要副产物为二苄醚。催化剂的苯甲醇和苯甲醚傅克烷基化反应结果和N2吸附-脱附等温线见图1。
由图1a可见,当反应时间为0.5 h时,ZS-550和ZS-Ce-550上苯甲醇的转化率分别为15.0%和73.3%,产物的选择性分别是87.1%和84.7%。实验结果表明,掺杂Ce会提高介孔SiO2-ZrO2的酸催化活性。
材料的比表面积会影响它的催化活性,一般而言,同等情况下,比表面积越大催化活性越高。由图1b可见,ZS-550和ZS-Ce-550的吸附平衡等温线均为Ⅳ型,有明显属于H2型的滞后环,证明材料具有介孔结构;从孔径分布也可以看出两者都有介孔结构。ZS-550的孔集中在5.5 nm,ZSCe-550的孔主要集中在6.8 nm,这可能是由于Ce的离子半径大于Si和Zr,从而使孔径变大,也间接证实了Ce进入了SiO2-ZrO2结构中。掺杂Ce后,介孔SiO2-ZrO2的比表面积有所增加,从218 m2/g(ZS-550)增至273 m2/g(ZS-Ce-550)。虽然比表面积有所增加,但不足以使催化活性提高近4倍,因此比表面积增大不是催化活性提高的主要原因。
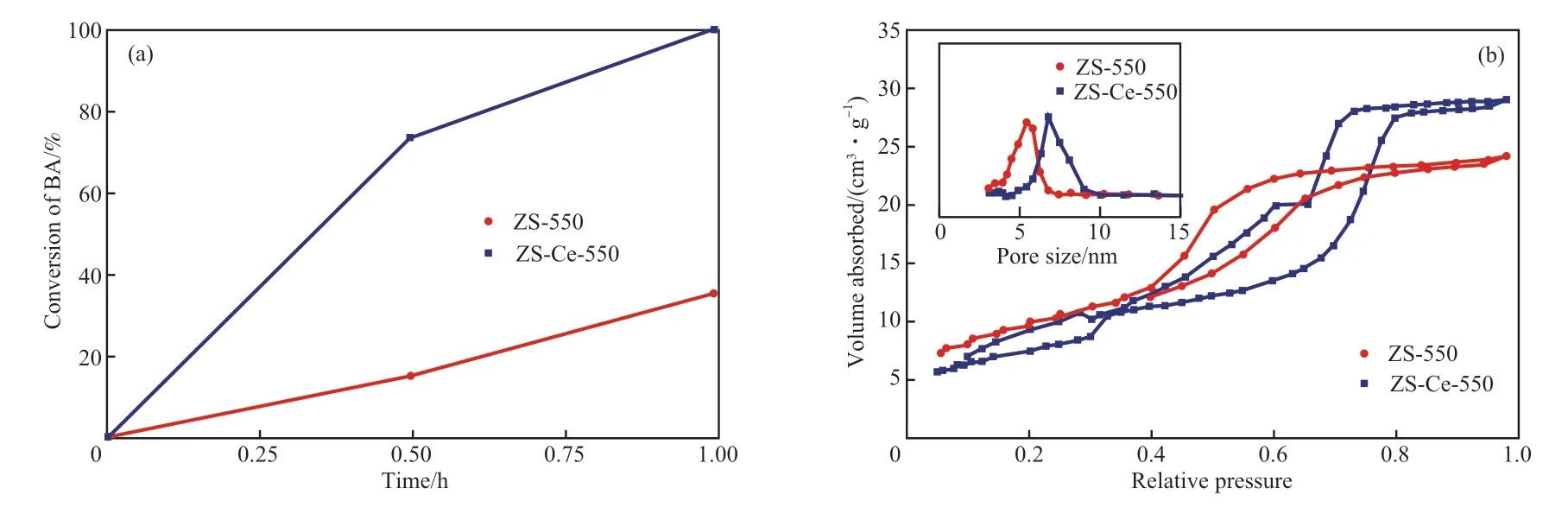
图1 催化剂的苯甲醇和苯甲醚傅克烷基化反应结果(a)和N2吸附-脱附等温线(b)Fig.1 Friedel-Crafts reaction results of anisole and benzyl alcohol(BA)(a) and N2 adsorption-desorption isotherms(b).Reaction conditions:154 ℃,0.1 g catalyst,5.445 mL anisole,0.518 mL BA.ZS-T:mesoporous SiO2-ZrO2 calcined at T ℃;ZS-Ce-T:mesoporous SiO2-ZrO2 doped with Ce,calcined at T ℃.
采用Py-FTIR对两种催化剂进行了表征,以研究两种催化剂活性有差异的原因。ZS-550和ZS-Ce-550催化剂的Py-FTIR谱图见图2a。由图2a可见,两种催化剂在1 611 cm-1和1 448 cm-1处有明显的吸收峰,是吡啶吸附在L酸位点上的峰,表明催化剂中含有L酸位点。此外,ZS-Ce-550催化剂在1 542 cm-1处还有一个明显的吸收峰,为B酸位点的特征峰[17],ZS-550催化剂的该吸收峰不明显。对应的吸收峰面积越大,酸含量越多,因此,ZS-Ce-550催化剂拥有更多的B酸位点。实验中吡啶脱附温度是150 ℃,所以催化剂中的酸位点是中强酸位点。
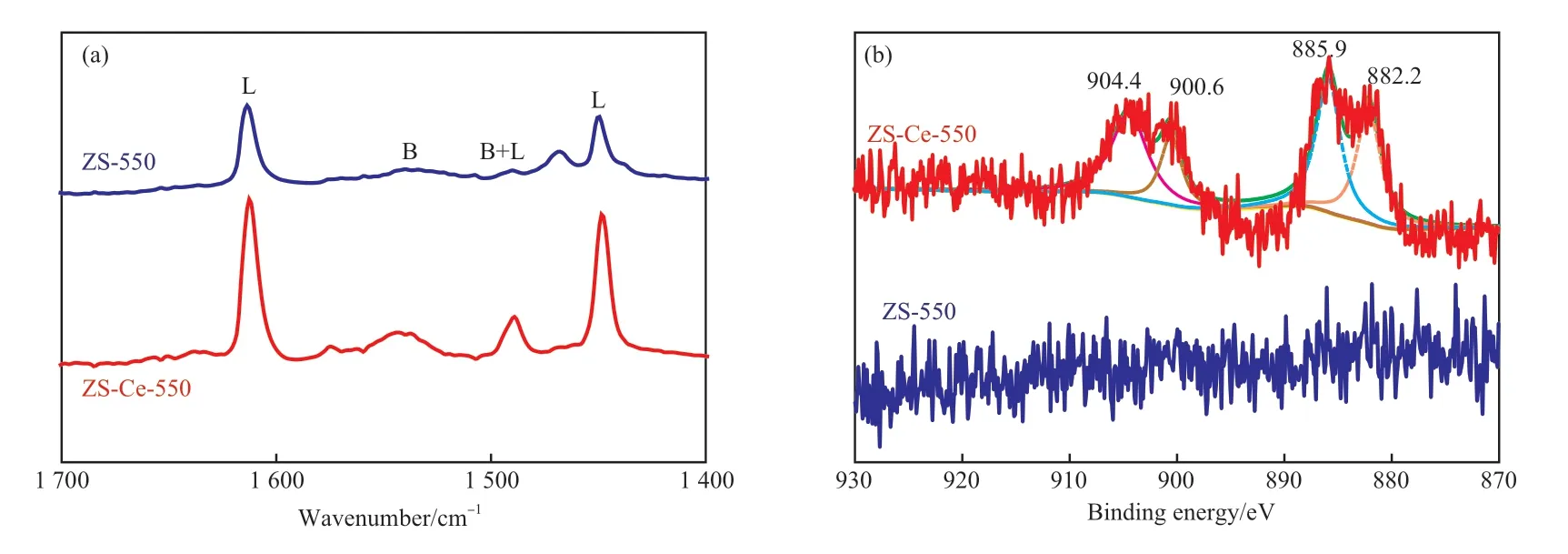
图2 ZS-550和ZS-Ce-550催化剂的Py-FTIR谱图(a)和XPS谱图(b)Fig.2 Py-FTIR(a) and XPS(b) spectra of ZS-550 and ZS-Ce-550 catalysts.
Ce的结构形态可能会对材料的催化活性起作用,复合金属氧化物中金属原子的价态和配位数对材料的酸性影响较大[18]。ZS-550和ZS-Ce-550催化剂的XPS谱图见图2b,图中给出的是Ce 3d的XPS谱图。从图2b可看出,ZS-550的XPS谱图中没有Ce的特征峰,说明材料表面没有Ce元素;ZS-Ce-550的XPS谱图中885.9 eV和904.4 eV分别对应Ce3+的3d5/2和3d3/2特征峰,882.2 eV和900.6 eV对应的是Ce4+的3d5/2和3d3/2特征峰[19-20],说明掺杂的Ce以Ce3+和Ce4+两种形式存在。由XPS表征结果可得Ce/(Zr+Si)的原子比为0.69,与理论值0.74接近,表明Ce基本全部进入到SiO2-ZrO2材料中。
催化反应结果表明,掺杂Ce会提高SiO2-ZrO2的酸催化活性;Py-FTIR表征结果显示,掺杂Ce会提高SiO2-ZrO2的酸性。Ce掺杂提高酸性的原因来源于两部分:1)根据XPS表征结果,Ce以Ce3+的形式进入SiO2-ZrO2,而Si和Zr都是正四价,按照Tanable模型理论,引入低价态的Ce3+会导致体系电荷不平衡,需要额外的质子来平衡体系电荷,引入的H+会产生更多的质子酸位点[21]。通过Tanabe的假设,将六配位Zr原子加入到SiO2中会引起电荷不平衡,若电荷为正则材料中的阳离子充当L酸位点;若电荷为负则需要表面的质子来平衡产生的负电荷,这时会产生酸位点。在ZS-550中,不平衡电荷为-2,具有一定的B 酸酸性。掺杂Ce后,Ce进入SiO2-ZrO2中,取代了部分Zr原子,产生的不平衡电荷从-2变到-3,增加的不平衡电荷需要更多的质子去平衡,因此B酸酸量增加(见图3)。2)Ce3+有较强的极化和诱导作用,会夺走O—H上的电子,使SiO2-ZrO2的酸性增强[22]。
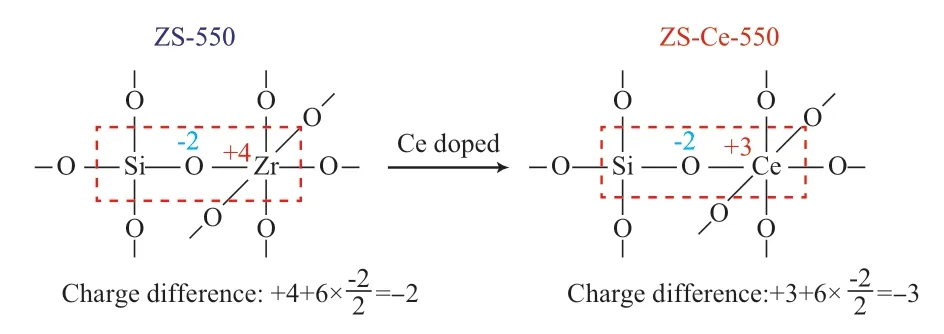
图3 Tanabe 模型中ZS-550和ZS-Ce-550中的不平衡电荷示意图Fig.3 Charge difference of ZS-550 and ZS-Ce-550 according to Tanabe model.
2.2 焙烧温度的影响
考察了焙烧温度对催化剂活性的影响,实验结果见图4a。由图4a可见,随着焙烧温度的升高,催化剂的活性逐渐提高。相比于ZS-Ce-450催化剂,ZS-Ce-550,ZS-Ce-650,ZS-Ce-750催化剂的活性提高较多,其中ZS-Ce-750催化剂的活性最高。
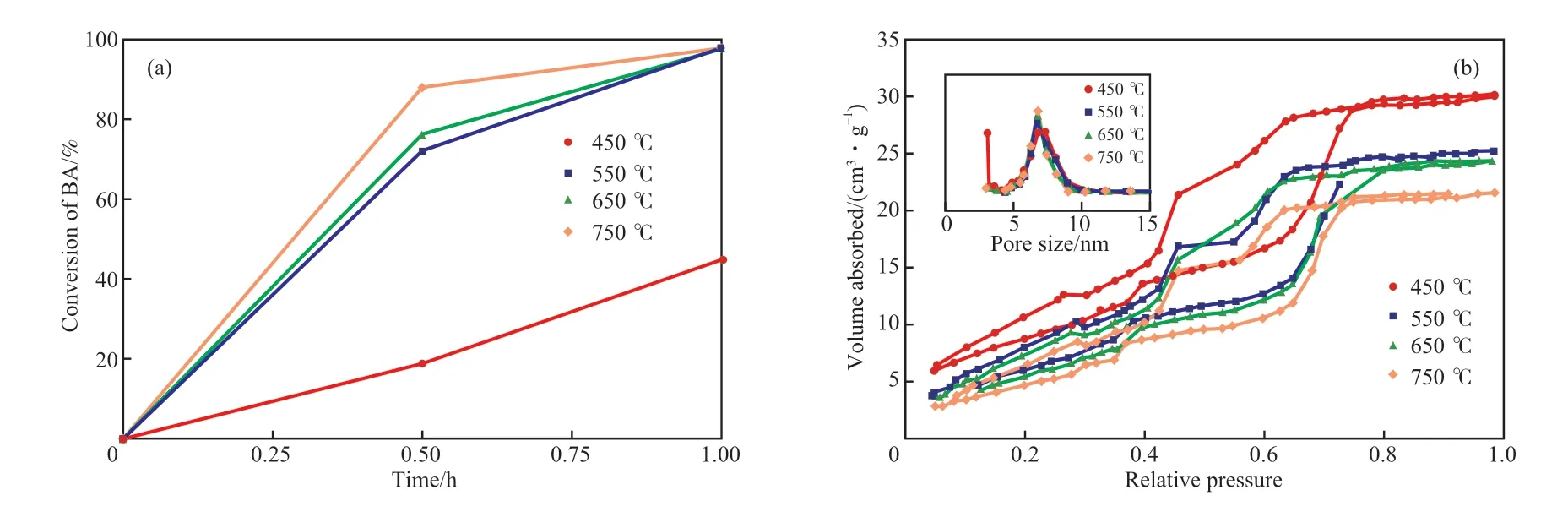
图4 不同焙烧温度下催化剂的活性(a)和N2吸附-脱附等温线(b)Fig.4 Catalyst activity(a) and N2 adsorption-desorption isotherms(b) of the catalysts calcined at different temperatures.Reaction conditions referred to Fig.1.
催化剂的活性与介孔结构和比表面积有很大的关联。不同焙烧温度下催化剂的N2吸附-脱附等温线见图4b。由图4b可见,焙烧温度升高并未破坏催化剂的介孔结构。随着焙烧温度的升高,比表面积减小:ZS-Ce-450(261 m2/g)>ZS-Ce-550(218 m2/g)>ZS-Ce-650(193 m2/g)≈ZS-Ce-750(196 m2/g)。一般情况下,升高焙烧温度会降低材料的比表面积和表面酸性(表面羟基减少,酸强度下降,酸量减少),从而使酸催化活性降低。但是根据催化活性的对比,焙烧温度升高导致ZS-Ce-T催化活性提高,这说明催化剂的比表面积不是影响催化活性的主要因素。
不同焙烧温度下催化剂的XPS谱图见图5。从图5可见,所有催化剂在881~886 eV和900~906 eV都有相应的特征峰,分别对应Ce的3d5/2和3d3/2。经过拟合,885.5 eV和904.7 eV左右处的峰归属于Ce3+,882.2 eV和900.6 eV左右处的峰归属于Ce4+,表明Ce是以Ce3+和Ce4+两种形式存在。因此,ZS-Ce-T催化剂的酸催化活性差异不是由Ce的价态变化引起的。
通过分析XPS表征结果,发现ZS-Ce-T催化剂表面Ce原子含量区别较大。ZS-Ce-T中Ce/(Zr+Si)的理论原子比为0.743,而XPS表征结果中Ce/(Zr+Si)的原子比随着焙烧温度的升高而增大。ZS-Ce-450的Ce/(Zr+Si)原子比为0.40,焙烧温度升至550 ℃和650 ℃时,Ce/(Zr+Si)原子比增加到0.69和0.63;当焙烧温度为750 ℃时,Ce/(Zr+Si)原子比高达0.85。这说明随着焙烧温度的升高,Ce3+不断地迁移到催化剂表面,最终富集在SiO2-ZrO2表面。催化剂表面Ce3+数目增加使酸性位点增多,反应大多在催化剂表面进行,因而随着焙烧温度的升高,酸催化活性提高。

图5 不同焙烧温度下催化剂的XPS谱图Fig.5 XPS spectra of the catalysts calcined at different temperatures.
3 结论
1)采用溶胶-凝胶法合成了介孔SiO2-ZrO2,SiO2-ZrO2掺杂Ce后,Ce进入SiO2-ZrO2中取代部分Zr原子,体系的不平衡电荷增大,增加的不平衡电荷需要更多的质子去平衡,因此B酸酸量增加,进而提高了它在苯甲醇和苯甲醚傅克烷基化反应中的催化活性。
2)Ce掺杂SiO2-ZrO2的酸催化活性随着焙烧温度的升高而增强,因为随着焙烧温度的升高,Ce3+不断在催化剂表面富集。当焙烧温度为750 ℃时,催化剂的活性最好。
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:04:56
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
环境科技(2017年6期)2018-01-17 08:58:33
当代化工研究(2016年2期)2016-03-20 16:21:18
纺织科技进展(2015年1期)2015-11-28 05:56:20
新高考·高一物理(2015年6期)2015-09-28 20:10:57
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
