
Fe@C包覆型催化剂的制备及其在费托合成反应中的应用
2020-08-21 14:57:06杨景麟林俊明陈文迪
石油化工 2020年7期
杨景麟,林俊明,陈文迪,岑 洁,姚 楠
(浙江工业大学 工业催化研究所 化学工程学院 绿色化学合成技术国家重点实验室培育基地,浙江 杭州 310032)
随着原油资源的枯竭和能源需求的增长,通过费托合成将煤、天然气或生物质衍生的合成气原料转化为液体燃料和其他化工产品的过程受到越来越多的关注[1-5]。在相关适用于费托合成的催化剂中,Fe基催化剂具有成本低和对合成气氢碳比适应性广等优点,因而极具工业化应用的潜力[6-7]。
Fe基催化剂主要分为熔铁催化剂、沉淀铁催化剂和负载型催化剂[8-9]。熔铁催化剂的比表面积、孔体积和活性较低,但可适用于高温反应。沉淀铁催化剂虽然具有较高的比表面积、孔体积和活性,但催化剂的机械强度较低,稳定性较差。相比于以上两种催化剂,负载型催化剂可以通过调节载体来提高催化剂的机械强度和分散度,并具有较高的比表面积和较大的孔体积。常规用于制备Fe基催化剂的载体主要是SiO2和Al2O3,但这两种载体与Fe前体物种之间易存在强相互作用,并生成难以还原的硅酸铁或铝酸铁,从而抑制了催化剂的还原与活化,进而影响费托合成反应性能[10-11]。而碳材料不仅具有可调的孔隙结构、较高的比表面积和易修饰的表面化学性质[12-13],且还可促进Fe基催化剂的还原与活性相的生成[14-15]。因此,碳材料在制备负载型Fe基催化剂领域具有广阔的应用前景[16]。同时,进一步的研究结果表明,与负载在碳材料表面相比,将Fe活性中心包覆在碳材料中形成包覆结构(即Fe@C)可抑制纳米粒子团聚或迁移,并通过限域效应显著影响Fe基催化剂的费托合成反应性能[17-22]。如碳纳米管(CNT)的π电子倾向于由内壁向外壁移动,这将削弱管内Fe2O3粒子的稳定性,提高催化剂的还原性。此外,由于独特的空间限制效应延长了活性中心与反应中间物的接触时间,所以管内活性中心对的产率是管外活性中心的2倍左右。
本文介绍了Fe@C包覆型催化剂结构、主要制备方法、费托合成反应性能及催化剂改性方面所取得的研究进展。
1 Fe@C催化剂的结构和制备方法
目前常用于制备催化剂的碳材料主要有活性炭、CNT和石墨烯等[23-26]。对于Fe@C包覆型催化剂,碳层既可以是球状的[27],也可以是管状的(如碳层由CNT组成)[24]。同时,碳层一般存在石墨化结构,且石墨化程度低的碳层具有较多的缺陷,有利于CO分子活化[14]。另一方面,碳层的厚度一般为几纳米到十几纳米,如Wang等[28]制备的Fe@C粒子中的碳层由2~5层石墨烯组成,厚度大约为1~2 nm。Long等[29]以金属有机骨架(MOFs)为前体,通过直接热解法制备的氮掺杂碳包覆型催化剂的碳层厚度为10 nm左右。过厚的外层结构可能会导致传质困难,影响催化剂的性能[30]。对于适用于费托合成的Fe@C包覆型催化剂,核一般由碳化铁等含铁物种组成[6,31-33]。虽然目前已经报道的制备Fe@C包覆型催化剂的方法有很多,但总体可归纳为一步热解法和后包覆法。
1.1 一步热解法
对于一步热解法,一般先将碳源和含Fe前体混合以形成混合物或配合物,然后再通过在惰性气氛下焙烧处理来合成Fe@C催化剂。在焙烧过程中,含Fe前体部分或完全分解,且在高温下发生碳热还原反应,铁氧化物被进一步还原。在该方法中,常用的碳源有葡萄糖和炭黑等。Tu等[34]通过调变Fe@C前体中葡萄糖的加入量以调控催化剂的粒径大小。表征结果表明,葡萄糖和铁物种的组合可形成Fe3O4@C纳米粒子结构,且增加葡萄糖含量有利于减少Fe3O4@C粒子的粒径,从而提供了更多的表面铁物种,这有助于铁氧化物的还原和铁活性中心的形成。Lu等[35]还以炭黑为碳源,研究了碳热反应中Fe@C包覆结构的形成过程。在焙烧催化剂时,铁前体受热分解为Fe2O3,然后炭黑将Fe2O3逐步还原成Fe3O4、FeO和Fe。在此过程中,C 2p轨道和Fe 3d轨道上未成对电子之间的吸引力,对碳壳层的自组装形成具有重要的作用。在碳热反应条件下,与铁纳米颗粒结合的碳原子将演变成最低能量构型,即碳原子在金属铁颗粒表面上沉淀,从而降低表面能,最后形成铁碳化合物。
在一步热解法中,受益于高比表面积、大孔隙体积以及结构和组合物的可调性,以MOFs为前体来制备金属催化剂的方法被广泛应用于各大领域中。现在可供选择的商用MOFs材料多种多样,这有利于研究者通过选择不同的MOFs材料达到制备不同催化剂的目的。与其他方法相比,利用MOFs来制备催化剂的过程更加简便,一般是将MOFs在惰性气氛中进行热解,使MOFs碳化为包裹Fe纳米颗粒的多孔碳基质。与普通制备方法相比,以MOFs为前体的Fe基催化剂具有高负载率。Wezendonk等[36]使用多种商用MOF材料制备了Fe@C催化剂,所得催化剂都表现出高Fe负载率(w)(36%~46%)。除此之外,Wezendonk等[37]还发现通过热解MOF材料所制备的Fe@C催化剂粒子的粒径为3.6~6.8 nm,且与Fe负载量无关。同时,调节相关条件可以有效控制催化剂的晶相[31]。以MOFs为前体所制得的催化剂的机械强度较弱,易在反应过程中破碎或粉化,进而容易使固定床反应器中的催化剂床层发生堵塞。为了解决这个问题,Oar-arteta等[21]在Fe-BTC(1,3,5-均苯三羧酸铁)MOF材料中加入一定比例的AlOOH作为黏合剂,研究AlOOH加入量对以MOFs为前体所得催化剂机械强度的影响。实验发现AlOOH的加入明显增加了催化剂的介孔结构,但在热解过程中从AlOOH释放的水易导致催化剂中铁物种的还原程度降低。为了评估AlOOH的加入量对催化剂机械强度的影响,研究者比较了不同Al掺杂量的催化剂38Fe@C,33Fe@C,25Fe@C,15Fe@C(xFe@C/Al,x代表Fe所占比例)复合材料在50 h反应期间内的稳定性,它们在反应器中塌陷的时间分别为3,22,50 h和未塌陷(50 h)。表明添加一定比例的AlOOH有助于增强催化剂的机械强度。
除了碳源及其含量外,不同的焙烧温度也会影响催化剂的性质。Cruz等[38]研究了不同温度(500,600,700 ℃)焙烧处理所得催化剂的晶相变化。研究结果表明,随着焙烧温度的升高,催化剂晶粒尺寸逐渐变大。同时,经过500 ℃焙烧的催化剂中仅存在α-Fe2O3晶相,而经过600 ℃和700 ℃焙烧的催化剂均产生部分Fe3C晶相。Tang等[15]通过谷氨酸与Fe物种的配位作用来合成高分散Fe配合物,并通过改变热解温度调变碳层结构。实验结果表明,碳层石墨化程度随着热解温度的升高而逐渐增加,这会提高导电性能,从而增强Fe与CO分子间的相互作用。Duan等[24]研究了不同焙烧温度对CNT包覆Fe粒子的影响。在经过600 ℃焙烧的催化剂中,一些纳米粒子被包裹在无序碳层中。随着热解温度升至750 ℃,可观察到无序碳层和竹状CNT结构,且随着热解温度的进一步升高,形成了更多的N掺杂CNT包覆的金属粒子。另外,被CNT封装的Fe纳米颗粒表面还存在一层石墨结构。这种由氮掺杂的碳纳米管(NCNT)和石墨层形成的双层包覆结构可以增加纳米颗粒的稳定性。与常见CNT的直径(小于50 nm)相比,所得NCNT的直径约为100~150 nm,具有更大的比表面积。
以上合成方法都是先通过形成较稳定的Fe—C混合物或配合物,再经过预聚合和高温焙烧得到Fe@C结构。虽然该合成过程和步骤较为简单,但是较难控制催化剂的颗粒尺寸、碳层厚度与缺陷程度。
1.2 后包覆法
与一步热解法相比,后包覆法首先进行碳层制备,然后再通过熔融、渗透等方法对Fe粒子进行包裹处理以制备Fe@C催化剂。这么做的目的是为了调控碳层厚度与缺陷,且有利于抑制焙烧过程中金属相的团聚,从而实现对含Fe粒子尺寸进行有效调控的目的。Kang等[32]首先制备了介孔碳CMK-3,然后将Fe(NO3)3与CMK-3通过物理研磨均匀混合,并在聚丙烯瓶中进行恒温老化,最后在CO气氛中进行焙烧,最终得到具有较纯Fe5C2晶相的催化剂Fe5C2@CMK-3。与普通碳材料相比,使用CMK-3碳材料促进了Fe5C2粒子的均匀分布(粒子平均直径仅为4.6 nm)。通过计算模拟,研究者还指出,CMK-3无定形碳上的Fe5C2粒子的CO解离能量势垒更低,电荷转移比单层石墨烯上的Fe5C2更高,这些因素都有利于提升催化剂活性。为了对催化剂的粒径进行调控,Teng等[6]制备了一种SiO2@RF(RF为间苯二酚-甲醛树脂)模板。然后,Fe通过超声-过量浸渍法掺入,模板中的SiO2通过碱洗除去。当使用150 nm直径的球状SiO2为模板并将前体置于500 ℃焙烧时,催化剂的颗粒大小为(7.3±1.6) nm。通过改变球状SiO2直径或焙烧温度可以将催化剂粒子的直径控制在7.3~12.1 nm范围内。虽然通过后包覆法可以有效控制催化剂的粒子尺寸,但该制备过程较复杂,难以在工业生产上大规模应用。
除了上述2大类制备方法,还有研究者提出其他新型制备方法。Tu等[39]通过简单的一步溶剂热法合成了Fe3O4@C,制备过程无需焙烧。Yu等[18]通过直接水热法制备了FexOy@C催化剂,库仑相互作用使FexOy纳米颗粒与小的碳质胶体结合,形成FexOy-in-C纳米棒,通过分子间脱水自组装成FexOy@C球体,过程简单高效,适用于多种糖与硝酸盐的组合。Wang等[28]通过电弧放电方法制备了石墨烯包覆的Fe基催化剂,与普通制备方法相比,以石墨烯为壳层的催化剂可在大气下存储3 d而不发生氧化,从而证明外碳层可以有效保护Fe纳米粒子免受氧化影响。虽然这些不同的方法最终都能得到Fe@C包覆结构,但也明显存在着制备过程复杂程度和成本不同等差异性。其中,一步热解法的过程和成本较低,但难以控制粒子的尺寸和碳层结构。相比较而言,虽然后包覆法有利于调控粒子尺寸,但过程较为复杂。
2 Fe@C催化剂在费托合成反应中的应用
Fe基费托合成催化剂的催化活性主要由碳化铁物种的性质(晶型、尺寸等)决定,并与铁氧化物前体的还原性、铁氧化物与载体的相互作用以及助剂等有关[6,19,35]。在制备Fe@C的过程中,碳材料种类和表面酸碱度、Fe—C之间的相互作用都会影响粒子结构(碳层形貌和厚度)、尺寸、机械强度,从而进一步影响传热、传质以及催化性能。
与负载型催化剂相比,包覆型催化剂的优点在于它的高稳定性。由于费托合成Fe基催化剂是结构敏感型催化剂[40],因此这种稳定性主要来源于包覆结构对催化剂尺寸的控制。Qin等[41]通过闪蒸热解和水热处理分别得到了Fe@C和Fe/C催化剂,并对它们的费托合成性能进行了比较。实验发现在350 ℃、2 MPa和H2/CO体积比为1的费托合成反应条件下反应100 h后,与新鲜催化剂相比,Fe/C催化剂的粒子增长了8 nm,而Fe@C催化剂仅增长4 nm。Lu等[35]在商用炭黑载体上浸渍了一定量的Fe,通过高温碳化和在沸腾HCl溶液中的回流酸洗,制备所得的催化剂具有明显的核壳结构。在310 ℃、1 MPa和H2/CO体积比为1的反应条件下,催化剂反应100 h后仍然保持87.5%左右的CO转化率,而且C5+以上产物选择性高达65%。这些催化剂的高稳定性可以归因于包覆结构的空间限域效应有效地抑制了烧结和粒子长大,从而增强了催化剂的稳定性。Wu等[25]比较了Fe@BCNNSs(BCNNSs:氮化硼碳纳米片)和浸渍法制备的Fe/BCNNSs的反应活性与稳定性。在320~360℃温度区间内,Fe@BCNNSs催化剂在反应过程中始终维持80%以上的CO转化率,而Fe/BCNNSs的CO转化率仅为8.7~12.3%。研究者进一步在320 ℃下对Fe@BCNNSs进行约1 000 h的长周期反应稳定性测试。实验结果表明,该催化剂的CO转化率仅下降5.5%左右,且仍然维持在80%以上。这可以归因于包覆结构有效地抑制活性中心的迁移和聚集,从而保证了反应稳定性。Hong等[42]采用CO气氛下热处理方法将通过水热反应制得的草酸铁二水合物立方体转化为Fe5C2@C纳米粒子,且形成介孔结构。其中,草酸铁二水合物与CO在一定温度热处理条件下发生化学反应形成Fe5C2活性相。新鲜催化剂未经过活化预处理步骤,直接在320 ℃、1.5 MPa和H2/CO体积比为1的条件下进行费托合成反应。该催化剂显示出较高的活性,与之相比,传统Fe/SBA-15催化剂需要进行复杂的活化预处理,且活化后催化剂的反应速率仅有1.0×10-4mol/(g·s)。
Zhu等[27]比较了碳包覆的FeMn(FeMn@C)催化剂和FeMn催化剂的反应性能。在280 ℃、2 MPa、H2/CO体积比为2的反应条件下,FeMn催化剂(CO转化率40%)具有比FeMn@C(CO转化率27%)更高的初始活性,这主要是由于碳层覆盖了部分活性中心造成的。但随着反应时间的延长,FeMn催化剂逐渐失活。在200 h内,CO转化率逐渐从40%降至20%。相比之下,FeMn@C催化剂由于碳层不可避免地覆盖一些活性位点,导致它的初期活性相对较低(CO转化率为27%),但碳层可以促进碳化物形成,并阻止粒子烧结,所以催化剂的CO转化率逐步提高到48%,且在随后的反应阶段中保持基本稳定。除了活性和稳定性之外,FeMn@C对于C2~C4烯烃的选择性高达40.6%,C2~C4产物的烯烷比为2.6。相比之下,FeMn催化剂的C2~C4烯烃选择性仅为20.2%,烯烷比为0.9。这是因为碳层阻止了低碳烯烃的再吸附,从而提高了FeMn@C催化剂的低碳烯烃选择性。
碳层对催化剂结构和反应性能的影响见表1。
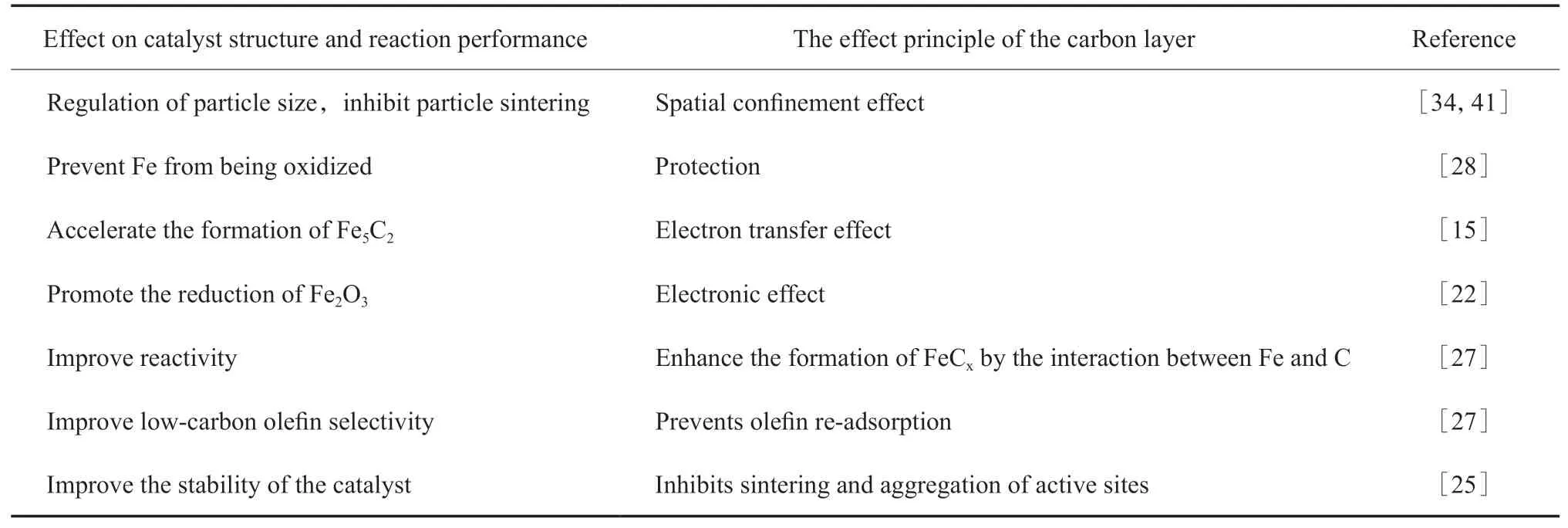
表1 碳层对催化剂结构和反应性能的影响Table 1 The effect of carbon layer on the catalyst structure and reaction performances
表1汇总了碳层对催化剂结构和反应性能的影响及其作用原理。从表1可以发现,碳层可以通过空间限域效应、电子传递效应或Fe与C间相互作用等影响催化剂的粒子尺寸、铁物种晶相等性质,进而提高催化剂的活性与稳定性、改变产物选择性。
3 Fe@C催化剂的改性
虽然Fe@C包覆结构极大地提高了Fe基催化剂的性能,但掺杂氮或钾可以进一步有效改变管状或球状碳包覆结构中的电子传输效果,进而影响催化剂表面对不同分子的吸附能力。因此,添加氮或钾助剂是改性该类催化剂的常用方法。
3.1 氮掺杂
对于碳材料负载型金属催化剂,通过氮掺杂可以改变碳材料的电子结构,进而促进共轭效应的产生和诱导电荷积聚,并传递到金属活性中心,从而改变活性中心对不同反应物分子的吸附。Williamson等[19]制备了氮掺杂的Fe@NCNT催化剂,并应用于逆水煤气-费托联合反应中。研究结果表明,与未掺杂的Fe@CNT催化剂相比,氮掺杂将Fe@NCNT催化剂的CO2转化率从48%提高到60.3%,因此掺杂氮有利于逆水煤气反应的进行。同时,Fe@NCNT对产物的选择性大大增加,这种趋势可能反映了Fe@NCNT催化剂对含偶极子的反应物(如CO,CO2)的亲和力增加,并增强了非极性产物(如短链烃产物)的脱附。Duan等[24]通过XPS表征不同温度处理后的催化剂中氮物种的存在形式。他们发现,掺杂到碳材料中的氮主要有吡啶型氮、吡咯型氮、吡啶-N-氧化物和石墨型氮这4种形式,且在不同的温度条件下可以进行转变。另外,当焙烧温度由750 ℃上升到1 050 ℃,氮掺杂量(x)从3.23%减少到1.55%,这表明在较高的热解温度下,大多数不稳定的氮物种都会分解。由于掺杂氮的种类较为复杂,所以较难阐明它对催化剂的促进作用。为了解决这一问题,Liu等[43]通过一锅法制备了仅含吡咯型掺杂氮的Fe基催化剂。实验结果表明,随着掺杂氮含量逐渐增加到2.1%(x),催化剂C5+选择性逐渐上升。他们认为这是由于较高吡咯氮含量的掺杂有利于C—O键强度的削弱和Fe—C键强度的增强,促进了CO的解离吸附,且催化剂表面活性中心吸附物种的变化使H2/CO体积比下降,从而使重质烃选择性增加。
除此之外,氮掺杂也会影响催化剂表面碱性。Shi等[44]研究了不同氮掺杂量对负载型Fe基催化剂的影响。研究结果表明,改变氮掺杂量并未明显改变CO转化率,但醇类选择性完全不同。未掺杂氮的催化剂的醇类选择性为20.2%。当氮含量到达1.3%(x)时,醇类选择性明显增加,最大值为27.2%。但具有2.4%(x)氮含量的催化剂对于醇选择性仅为21.4%,这表明掺入适量的氮助剂有利于获得最高的醇选择性。这种醇类产物选择性的改变是因为氮助剂改善了载体上的表面碱性,从而增加催化剂上CO化学吸附来影响反应产物中的总醇选择性。
3.2 钾掺杂
对于Fe基费托合成催化剂,添加适量的K助剂不仅可以减弱C—O和Fe—H化学键的强度,且能增强Fe—C键的强度,从而有利于生成长链烃[45-46]。常见的引入K助剂的方法是通过浸渍法[46-47],但通常会导致K助剂分散不均匀,且不能准确地控制K助剂的含量。与普通浸渍法相比,Zhou等[7]以柠檬酸钾为前体,将K助剂直接带入碳纳米片负载的Fe基催化剂体系中,通过柠檬酸钾的碳化作用可以使K均匀分布在整个碳材料上。在相同的反应条件下,相较于非掺K的催化剂,该催化剂具有更好的低碳烯烃选择性。
对于K助剂,除了分布均匀性以外,K助剂的含量也会影响催化剂的性能。Tian等[14]通过在硝酸铁-葡萄糖前体中加入不同量的K助剂来研究K含量对Fe@C催化剂的影响。他们发现,K的掺杂可以明显减小含Fe纳米粒子的粒径,这将大大增加活性中心与反应物分子间的接触面积。同时,Raman表征结果表明,未掺杂K助剂的催化剂的D带与G带之比(ID/IG)为0.91。随着K含量的增加,试样的ID/IG首先增加到1.13,然后略微降低。该结果表明添加K助剂改变了碳材料的结构,形成了更多的缺陷,这有助于反应过程中CO分子的缔合吸附和解离吸附。在另一方面,K助剂还会影响催化剂中Fe物种的还原与碳化。Niu等[48]研究发现,掺杂K助剂会抑制α-Fe2O3还原为Fe3O4,这是因为钾助剂存在时会增加Fe和O的电子密度,从而增强Fe—O键。Pendyala等[49]研究发现掺K提高了Fe剂催化剂的碳化速率。因此,K助剂具有提高Fe基催化剂碳化性能、增加碳材料缺陷、调控Fe纳米粒子粒径等功能。同时,K助剂的含量和分布均匀度也对催化剂的性能产生重要的影响。
4 结语
与普通Fe基催化剂相比,Fe@C催化剂可通过限域效应及Fe-碳层间的协同效应抑制纳米粒子团聚、迁移,促进Fe基催化剂的反应活性、稳定性和产物选择性。同时,通过N和K等助剂的修饰,进一步调整金属活性中心的电子性质和碳层结构,优化不同分子在活性中心表面的吸附、解离,从而提升催化剂的反应性能。目前通过一步热解法、后包覆法等可制备Fe@C催化剂,但这些制备方法都有优缺点。一步热解法具有制备过程相对简单的特点,所制得的Fe@C催化剂在反应过程中具有较好的活性和稳定性,但该方法难以调控碳层的厚度和缺陷结构。相比之下,后包覆法制备过程较为复杂,但能够更有效地调控碳层结构以影响反应物和中间产物的传质、吸附和反应,进而提高费托合成的反应活性和产物选择性。因此,在Fe@C催化剂制备和应用研究领域,今后的研发重点是结合一步热解法和后包覆法等方法的优点,开发获得能准确调控Fe@C核壳粒子纳米结构(缺陷、尺寸)和电子性质的新合成方法,从而获得目标产物选择性高、稳定性好的Fe基催化剂。
猜你喜欢
山西化工(2022年7期)2022-11-06 11:05:22
天津医科大学学报(2021年1期)2021-12-05 11:11:05
物理学报(2019年1期)2019-01-25 09:53:52
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
载人航天(2016年3期)2016-06-04 06:08:42
陶瓷学报(2015年4期)2015-12-17 12:45:02
化工进展(2015年6期)2015-11-13 00:27:28
金属加工(热加工)(2014年1期)2014-10-08 11:38:22
茶叶通讯(2014年2期)2014-02-27 07:55:40
河北医科大学学报(2011年3期)2011-03-25 10:15:59
