
介孔NiS2/S-g-C3N4的制备及其光催化产氢性能研究*
2020-08-03 03:29苏扬航韩亚翔陈绪兴
功能材料 2020年7期
刘 凯,苏扬航,韩亚翔,陈绪兴,高 云,李 荣,2
(1. 湖北大学 材料科学与工程学院, 武汉 430062; 2. 结构化学国家重点实验室, 福州 350002)
0 引 言
随着人类社会的快速发展,以化石能源的枯竭所带来的能源问题日益凸显,利用半导体光催化分解水产氢被认为是最有希望解决能源危机的技术之一。1972年Fujiashima等人发现TiO2可以光电催化分解水制氢,从此拉开了光催化分解水制氢的序幕[1]。随后针对TiO2光催化分解水制氢活性的优化,科研工作者展开了大量研究[2-8]。由于TiO2较大的禁带宽度(3.2 eV)使其仅对太阳光谱中的紫外光具有响应,而紫外光仅占太阳光谱中能量约5%,导致其光能利用率低,实际应用受到了极大限制[9]。2009年,福州大学王心晨教授发现石墨烯相氮化碳(g-C3N4)在可见光下可以分解水制氢[10],这种化学性质稳定,易于制备、成本低廉的具有类石墨烯层状结构的材料在光催化领域迅速引起了广泛的关注[11]。g-C3N4的禁带宽度约为2.7 eV,可以吸收太阳光谱中λ≤460 nm的蓝紫光,相较于传统的金属氧化物半导体光催化剂,其光能利用率较高,然而较低的比表面积、较高的载流子复合率及较低的表面反应速率[12-14],导致纯g-C3N4表现出非常低的光催化产氢性能,严重制约了g-C3N4的实际应用。
针对上述问题,科研工作者展开了大量研究。对g-C3N4进行掺杂,以调节g-C3N4的光学、电子能带结构等特性。例如:2009年,王心晨教授等报道在g-C3N4框架中掺杂金属元素Zn2+和Fe2+,并且发现随着掺杂金属元素含量的增加,金属/g-C3N4复合材料相比g-C3N4扩大了其对太阳光谱的光吸收范围[15]。2010年,成会明教授等制备了S掺杂g-C3N4,优化了催化剂的电子结构,价带的负移增加了光生空穴的氧化能力[16]。2011年,王心晨教授等又发现除了Zn2+、Fe2+离子外,其他过渡金属阳离子Ni2+、Cu2+、Co3+和Mn3+等,在不破坏g-C3N4主体结构的前提下,都可以掺入g-C3N4的框架中,并且表现出相似的效应[17]。2012年,陈亦琳教授等通过简单的H2O2水热处理合成了O掺杂g-C3N4,吸收带边移动至498 nm,有效地增强了可见光响应[18]。综上所述,元素掺杂可以明显拓展g-C3N4的吸收光谱。理论上单层g-C3N4具有较大的比表面积,然而通常制备的块状g-C3N4存在严重的层堆积现象,导致其比表面积较低,光生电子和空穴还没有迁移到催化剂表面参与反应就在催化剂体内复合,载流子复合率较高。针对这个问题,研究者采用剥离法来制备较大的比表面积的g-C3N4纳米片。例如:2012年,成会明教授等在空气中对块状g-C3N4进行热氧化剥离,获得了厚度约为2 nm,比表面积为306 m2/g的g-C3N4纳米片,其电子传输能力提高并且载流子寿命增加,光催化产氢性能得到了提高[19]。2013年,王心晨教授等通过简单的液相超声剥离块状g-C3N4得到g-C3N4的纳米片,纳米片的厚度约为2 nm,比表面积更是高达384 m2/g,在可见光下其光催化产氢性能提高了大约9倍[20]。同时,将两种具有合适导带和价带位置的报道提构成异质结,利用异质结的内建电场来促进电荷转移和分离,也是一种有效的方法。例如:2011年,朱永法教授等制备了ZnO/g-C3N4杂化纳米材料,在紫外线照射下光催化活性增加了3.5倍[21]。2011年,John T.S. Irvine教授等发现SrTiO3-C3N4复合光催化剂在可见光产氢速率达到440 μmol/(h·g),纯g-C3N4的光催化性能提高很多倍[22]。尽管通过剥离、构建异质结可以促进光生载流子分离,但是g-C3N4表面较低的反应速率又大大抑制了光催化反应速率,负载助催化剂也是一种行之有效的方法,例如:2009年,王心晨教授等用光化学沉积的方法,在g-C3N4上沉积不同的贵金属纳米颗粒(Rh、Ru、Pd、Pt、Au等),并比较了其在可见光下分解水制氢的性能,发现Pt是最好产氢助催化剂,光催化产氢效率提高了7倍[23]。2010年,Markus Antonietti教授等通过沉积-沉淀、光沉积和浸渍等方法在g-C3N4上沉积金纳米颗粒,使其光催化活性得到明显提升[24]。然而由于贵金属的昂贵价格,导致其不可能进行大规模使用。过渡金属硫化物由于其独特的光学和电学性质[25],H2在过渡金属硫化物表面的吸附自由能接近于零[26],被认为是一种理想的产氢助催化剂,可有效的提高g-C3N4的光催化产氢活性。例如:2013年,王心晨教授等在g-C3N4中引入了具有良好电催化产氢活性的MoS2,显著提高其光催化活性[27]。2014年,方晓明教授等通过一种简单的原位无模板离子交换工艺制备了NiS/g-C3N4复合光催化剂,并在光催化析氢测试中,最佳性能的样品g-C3N4/NiS-1.5%(摩尔分数)与2.0%(质量分数)Pt/g-C3N4的产氢速率几乎接近,NiS作为助催化剂可以有效的促进g-C3N4光生载流子的分离,从而提高光催化产氢活性[28]。
目前,经过科研工作者的努力,g-C3N4光催化产氢性能得到了显著提升,然而上述针对g-C3N4光催化活性的改善方法依然还存在一些缺点。例如:剥离制备的g-C3N4虽然具有超大的比表面积,但是其制备方法的产量极低,并不利于大规模制备。所制备的异质结界面接触不够良好,导致光生载流子分离效率依然有待进一步提高。同时,仅仅采用元素掺杂、形貌调控、构造异质结和负载助催化剂等改性方法中的一种或是两种方法的结合,其改善能力有限。如何采用一种经济、简单的制备方法将上述所有改善方法集于一体以进一步提升g-C3N4的光催化产氢性能依然是一个挑战。
鉴于上述考虑,本文中我们利用介孔SiO2作为硬模板,采用CH4N2S和Ni(CH4N2S)4作为前驱体,通过一步退火法成功制备了介孔S-g-C3N4负载NiS2复合光催化剂。利用介孔材料的有效传质效应;同时利用Ni-S配位原子层作为S-g-C3N4和NiS2的过渡连接层,确保了S-g-C3N4和NiS2间光生载流子的高效传输;最后利用非贵金属NiS2作为助催化剂来降低析氢过电位,促进H+的电催化还原,这为获得高效的g-C3N4基光催化产氢材料提高了保障,使其在可见光下(λ≥420 nm)的产氢性能达到141.33 μmol/(g·h),是纯g-C3N4的46倍。然后通过XPS、稳态/瞬态荧光光谱、电化学阻抗谱、线性伏安曲线、瞬态光电流对其光催化活性提升机理进行了进一步探究。
1 实 验
1.1 x-NiSCN复合光催化剂的制备
以CH4N2S和Ni(CH4N2S)4为前驱体,通过热缩聚的方法原位制备了具有不同NiS2含量的x-NiSCN纳米复合材料。具体实验步骤如下:首先,将5 g的粒径约为20 nm的SiO2纳米微球分散于100 mL去离子水中,在剧烈搅拌下加入10 g硫脲和一定比例的四水合乙酸镍,并搅拌30 min。其次,将所得溶液加热至100 ℃进行蒸发。待水分蒸干后将所得到的固体粉末在60 ℃干燥12 h,并进行充分研磨。再次,将研磨后的固体粉末置于带盖的刚玉坩埚中,在N2氛围下525 ℃退火,保温2 h,升温速率为5 ℃/min。最后,将退火后的样品分散在2M NaOH溶液中以刻蚀掉SiO2模板,并通过离心和抽滤洗涤得到最终样品,将其标记为x-NiSCN,其中x代表硫脲和乙酸镍的质量比。采用上述相同的制备方法,分别以采用尿素和硫脲作为原料,但不加入四水合乙酸镍制备了g-C3N4(标记为g-C3N4)和S掺杂的g-C3N4(标记为SCN)样品。
1.2 催化剂的表征
通过粉末X射线衍射仪(D8-Advance,X射线源Cu靶Kα射线,λ=0.15418 nm)和傅立叶红外光谱仪(Spectrum One)表征样品的物相结构于表面基团;使用分光光度计(日本SHIMADZU UV-3600)测试样品的U紫外-可见漫反射吸收光谱(UV-Visible DRS);使用FESEM(蔡司SIGMA 500)、TEM(Tecnai G20)观察样品的形态;利用X射线光电子能谱(XPS,ESCALAB 250Xi)对样品的组成及化学态进行表征,利用样品表面上污染碳的284.8 eV C 1s峰对所有结合能进行校准;利用荧光光谱仪(Perkin Elmer LS55)测试样品的瞬态/稳态吸收光谱,采用375 nm氙灯作为激发光源。
1.3 光催化产氢测试
光催化产氢实验采用北京中教金源的CEL-SPH2N光催化活性评价系统进行测试。采用超声法将10 mg光催化剂分散在50 mL水溶液(20 vol%三乙醇胺)中,加装420 nm滤光片的300 W氙灯作为光源,每隔60 min进行一次取样并利用气相色谱仪(GC7920,以N2为载气,TCD)分析H2的生成量。
1.4 光电化学测试
使用辰华电化学工作站(CHI 600E)的三电极系统进行光电化学测试,以Pt丝电极作为对电极,Ag/AgCl电极作为参比电极,在导电表面上涂有样品的FTO玻璃作为工作电极。工作电极制备过程如下:首先,将10 mg样品分散在0.5 mL DMF中,将其充分超声分散;然后,滴到FTO玻璃的导电表面上,最后,将获得的工作电极在50 ℃下加热约3 h,以蒸发残留的溶剂。光源为300 W氙灯,电解液为0.5 mol/L Na2SO4水溶液。在瞬态光电流响应和电化学阻抗谱(EIS)测试中。在0.1~106Hz的频率范围内以5 mV的振幅记录EIS谱线。在线性扫描伏安法(LSV)测量中,扫描范围为-0.65~-1.35 V(相对于Ag/AgCl,pH=6.6)。

图1 x-NiSCN复合材料制备示意图Fig 1 Schematic illustration for the fabrication of x-NiSCN composites
2 结果及讨论
2.1 形态和微观结构
所制备样品的形貌如图2所示。从图2(a)的SEM可知,SCN样品为褶皱的片状,图2 (c)的TEM图进一步显示该片层的厚度为纳米级。从图2(b)可知,在负载NiS2后,100-NiSCN样品仍然保持褶皱的片状,并且表面没有发现明显的NiS2大颗粒,图2 (d)的TEM图进一步显示NiS2纳米颗粒的尺寸大约为20 nm左右,并且较为均匀负载在SCN片上。

图2 SCN和100-NiSCN的SEM和TEM图Fig 2 SEM and TEM images of SCN and 100-NiSCN
2.2 物相结构与表面化学状态
所制备样品的XRD图谱如图3 (a)所示,g-C3N4和SCN都在2q为12.7和27.3°位置出现了明显的衍射峰,分别对应g-C3N4的(100)和(002)晶面。随着乙酸镍添加量的增加,SCN特征峰强度逐渐降低但并没有消失,说明其结构依然得以保存,而NiS2的特征峰的强度逐渐增强,并且随着乙酸镍含量的增加,特征峰的强度逐渐增加。对其基团进一步进行了FT-IR表征,如图3 (b)所示。g-C3N4,SCN和x-NiSCN均出现了位于1 100~1 700 cm-1处的CN杂环的特征峰和815 cm-1处的s-三嗪单元的尖锐特征峰,以及3 000~3 600 cm-1处的N-H特征宽峰。除此之外,在x-NiSCN中并没有观察到其他的特征峰,进一步说明NiS2的负载并没有破坏g-C3N4的本征结构。

图3 g-C3N4、SCN和x-NiSCN的XRD图和(b) FT-IR图谱Fig 3 XRD patterns and FT-IR spectrum of g-C3N4, SCN and x-NiSCN
通过X射线光电子能谱(XPS)研究了样品的表面化学性质。所有图谱的结合能都利用284.8 eV处出现的污染碳源的C 1s峰进行校准。图4 (a)是NiS2、SCN和100-NiSCN的C 1s高分辨图谱,NiS2中只含一个位于284.8 eV的峰,归属于表面污染碳的C-C键。在SCN样品中,碳有3个峰,分别位于284.8、286.3和288.0 eV,分别对应于碳C—C键、S掺杂到g-C3N4结构中的C—S键和g—C3N4中三嗪的N—CN2结构中的碳。100-NiSCN的C 1s谱图类似于SCN,但是N—CN2结构中的碳结合能向高移到288.2 eV,而C-S键的结合能向低移到285.9 eV。图4 (b)是SCN和100-NiSCN的N 1s高分辨图谱,对于SCN,N 1s图谱有4个峰,分别位于398.3、400.1、401.1和404.3 eV,它们分别是CN—C、(C)3-N、N-H中的N以及N的π激发。在100-NiSCN的中发现CN—C和N的π激发分别向高结合能移动到398.7和404.8 eV,而(C)3—N、N—H则向低移动至399.4和400.7 eV。图4(c)是NiS2,SCN和100-NiSCN的S 2p的高分辨图谱。SCN样品中的两个峰,分别位于162.3和167.8 eV,它们分别是由硫掺杂的g-C3N4材料中的硫取代氮而形成的C—S键以及材料表面的S-O键。NiS2中的3个峰,位于162.8和164.0 eV的是与Ni2+离子形成二硫化物的还有在168.4 eV为NiS2表面S-O键。在100-NiSCN样品中值得注意的是,Ni-S中的两个峰都向低结合能移动到162.5和163.7 eV,而在g-C3N4中掺杂的C-S键向高结合能移动到164.3 eV。图4 (d)是NiS2和100-NiSCN的Ni 2p的高分辨图谱。在NiS2中,854.0和871.4 eV分别是Ni 2p 3/2和Ni 2p 1/2,显示为Ni2+状态,857.8和877.9 eV为Ni的卫星峰。而在100-NiSCN中,所有的峰都向高结合能方向移动,Ni 2p 3/2和Ni 2p 1/2移动至855.7和873.4 eV,卫星峰移动至862.1和879.5 eV。
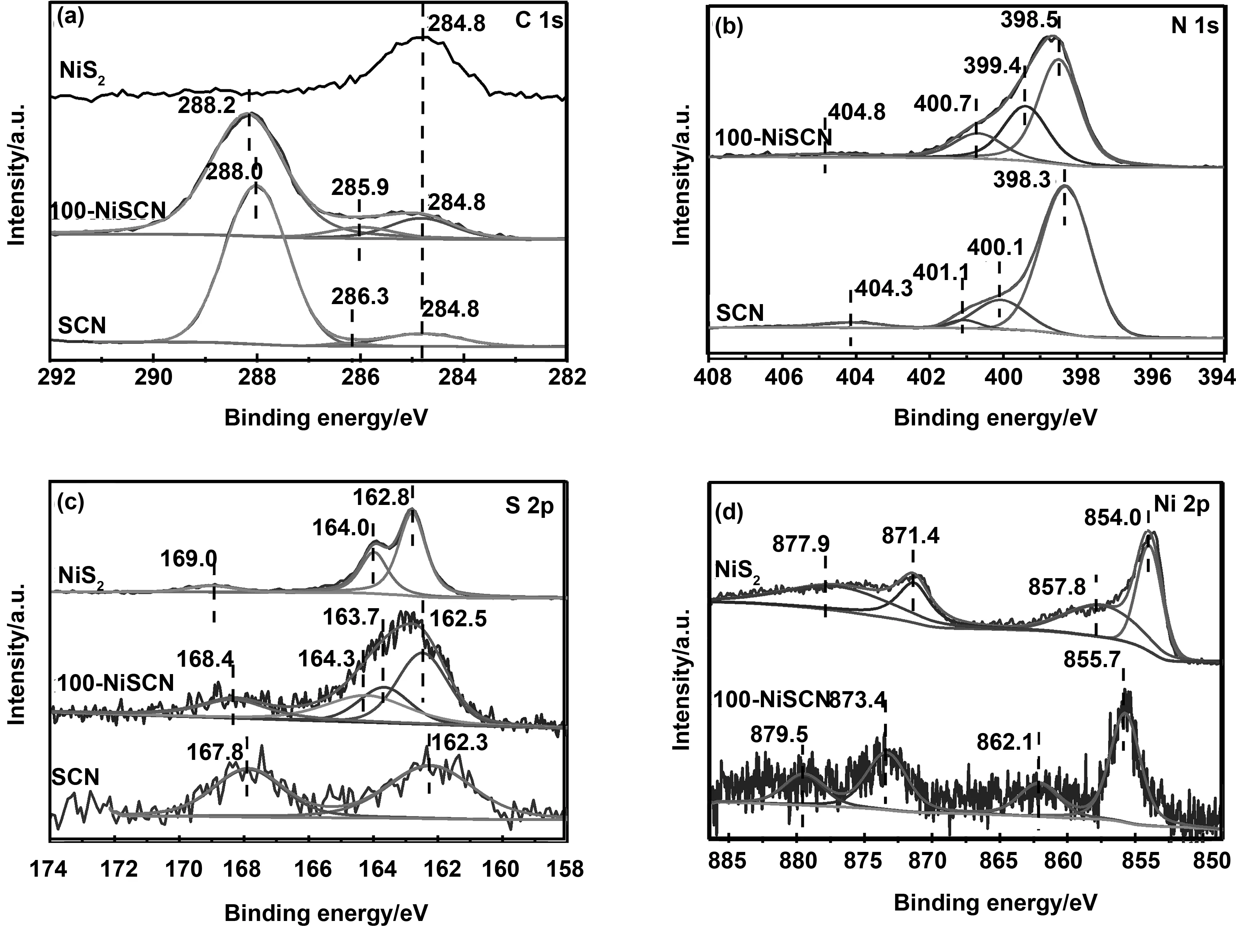
图4 SCN、NiS2和100-NiSCN的XPS高分辨能谱Fig 4 High-resolution XPS spectra of SCN, NiS2 and 100-NiSCN
2.3 光学吸收
通过紫外-可见漫反射吸收光谱来研究样品的光吸收特性,如图5(a)所示。利用硫脲作为前驱体制备的SCN的吸收带边,相对于g-C3N4吸收带边出现了的红移,禁带宽度进一步较小,从而提升了太阳光谱的利用率。具有不同NiS2含量的x-NiSCN复合样品均显出类似SCN的吸收边,这表明SCN的结构得以保存。但是x-NiSCN复合样品在可见光区域(500~800 nm)的吸光度大大提高,并且随着NiS2含量的增加,吸光度逐渐增大,这可以归因于黑色的NiS2较宽的光谱吸收。
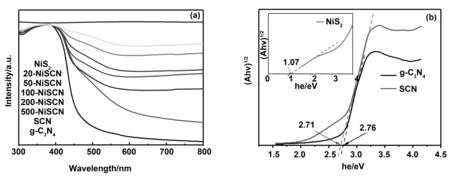
图5 g-C3N4,SCN,NiS2和x-NiSCN样品的紫外-可见漫反射吸收光谱和SCN和NiS2对应的Kubelka-Munk图Fig 5 UV-visible DRS of g-C3N4, SCN, NiS2 and x-NiSCN and Kubelka-Munk plots of SCN and NiS2
2.4 光催化产氢性能
g-C3N4、SCN和x-NiSCN的光催化产氢性能如图6所示。正如前言所述,由于较高的载流子复合效率和滞缓的表面反应速率,g-C3N4表现出微弱的光催化产氢性能,其产氢速率仅为3.05 μmol/(g·h)。虽然S元素的掺杂使SCN在可见光区的吸收增加,导致其光催化产氢速率提升到了5.21 μmol/(g·h),但由于其滞缓的反应动力学导致光催化产氢性能依然较低。在负载了NiS2之后,其光催化产氢性能得到了显著的提升,其中100-NiSCN样品表现出最高的光催化产氢性能,其产氢速率为141.33 μmol/(g·h),分别为g-C3N4和SCN的46和27倍。结果表明,S元素掺杂和助催化剂NiS2的引入可以有效的提高g-C3N4的光催化产氢性能。
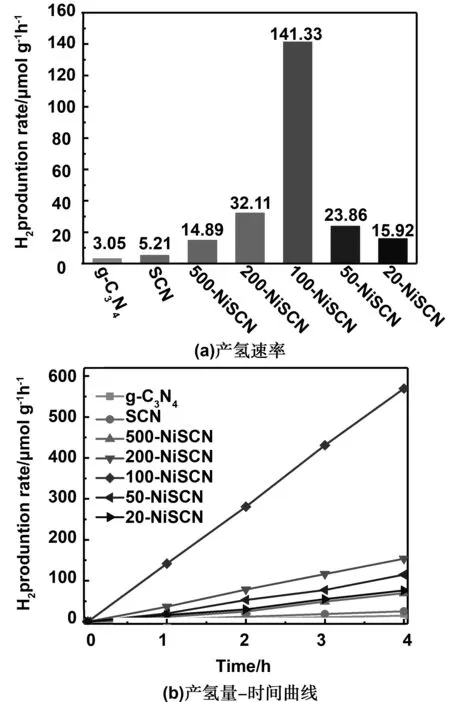
图6 g-C3N4、SCN和x-NiSCN复合材料的光催化产氢性能Fig 6 Photocatalytic hydrogen production performance of g-C3N4, SCN and x-NiSCN
2.5 光催化机理研究
为了进一步揭示光催化产氢性能提升的原因,对所制备样品进行了稳态/瞬态荧光光谱、电化学阻抗谱、线性伏安和瞬态光电流表征。如图7(a)所示,与之前报道一样,SCN样品在400~600 nm处有一个较宽的发射峰,为带间跃迁发射峰。在100-NiSCN的样品中,由于NiS2的引入,导致其发光光谱强度明显减弱,从而说明负载NiS2可显著降低光生载流子的复合效率。在图7 (b)的瞬态荧光光谱表征中,SCN样品光生载流子的平均寿命为1.54 ns,而负载NiS2之后,其光生载流子的平均寿命提升到1.80 ns,进一步证实了负载NiS2可促进光生电子-空穴的分离效率。从图7(c)所示的电化学阻抗谱中,可知SCN样品具有较大的电阻,而NiS2具有良好的导电性,随着NiS2的引入,SCN的导电性得到明显的改善,这有利于光生载流子的传输。从图7(d)中的线性伏安曲线可知,对于电催化析氢反应,SCN具有较大的过电势,这是由于其滞缓的反应动力学所致。而NiS2表现出良好的电催化析氢性能,可见其为一种良好的产氢助催化剂。因此,在引入NiS2后,SCN的电催化析氢性能得到了明显改善。上述实验结果表明NiS2的引入可显著提升SCN中光生电子与空穴的分离、运输,并有效改善其滞缓的反应动力学,从而可预测NiS2的引入可有效提高其光电催化性能。我们利用瞬态光电流进行了验证,如图8所示,相比于单纯的SCN和NiS2样品,100-NiSCN样品的光电流得到了显著提升,从而证实了上述预测。
通过上述结构表征及机理研究,可知100-NiSCN具有良好的光催化产氢性能的主要原因是:(1)利用SiO2作为硬模板合成介孔材料,提升了催化剂的传质能力;(2)S元素的掺杂拓展了g-C3N4对太阳光谱的吸收;(3)NiS2与SCN良好的界面接触促进了光生载流子的分离;(4)助催化剂NiS2的引入改善了g-C3N4本身滞缓的反应动力学,降低了其产氢过电位。
3 结 论
利用纳米SiO2作为硬模板,采用CH4N2S和Ni(CH4N2S)4作为前驱体,通过简单的退火法成功制备了介孔S-g-C3N4负载NiS2复合光催化剂。在可见光下 (λ≥420 nm),100-NiSCN样品的产氢速率可达141.33 μmol/(g·h),是纯g-C3N4的46倍。然后我们利用XRD、SEM、TEM、UV-visble吸收光谱、FT-IR、XPS、稳态/瞬态荧光光谱、电化学阻抗谱、线性伏安曲线、瞬态光电流对其结构与形貌、表面状态、光催化反应机理进行了详细研究,揭示了其光催化产氢性能得以提升的原因。该研究为制备高效的g-C3N4基光催化剂提供一种简单、高效、经济的设计思路,并为其进行大规模工业化生产及应用提供了理论基础。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
物理学报(2022年6期)2022-03-30
石油化工高等学校学报(2021年3期)2021-07-15
河南科学(2020年7期)2020-09-10
物理学报(2020年16期)2020-08-29
化工设计(2020年2期)2020-05-01
科技风(2018年9期)2018-05-14
郑州大学学报(理学版)(2017年1期)2017-04-07
化工管理(2017年25期)2017-03-05
