
特发性肺纤维化与细胞微环境之间关系的研究进展
2020-07-09 08:32孙梦阳王新华
中国医学科学院学报 2020年3期
叶 威,孙梦阳,王新华
浙江中医药大学第一临床医学院,杭州 310053
特发性肺纤维化(idiopathic pulmonary fibross,IPF)是一种以细胞外基质(extracellular matrix,ECM)过度产生和沉积,及由肺泡上皮细胞慢性损伤后的异常修复所导致的,以肺组织异常重构为特征的致命性间质性肺疾病[1]。IPF的发病机制尚未明确,目前认为其演变可分为以下3个过程:(1)易感过程:包括遗传突变、环境暴露和衰老等过程,这些过程使个体易患肺纤维化;(2)激活过程:转化生长因子-β(transforming growth factor-β,TGF-β)活化、纤维细胞募集、上皮-间充质转化(epithelial mesenchymal transition,EMT)和未折叠蛋白反应(unfolded protein response,UPR)的活化,促进纤维化的进展;(3)进展过程:如病理成纤维细胞分化、基质沉积和重塑、基质刚度增加以及成纤维细胞和上皮细胞内的促纤维化表观遗传学变化[2]。在已知/未知的因素下,持续的间充质细胞活化和基质重塑,使受损的肺上皮细胞重新编码,跳过前两个阶段,产生过量的ECM沉积,形成纤维疤痕,最终导致肺结构和功能丧失(图1)。本文将从细胞调节因子、间充质细胞、ECM、UPR 4个方面来阐述IPF发病机制与细胞微环境之间的最新研究进展。
细胞调节因子在IPF发病中的研究进展
TGF-βTGF-β是最重要的致纤维化因子之一,能促进成纤维细胞的增殖,刺激结缔组织合成并抑制其降解。纤维化是一个非常复杂的过程,其中 Smad信号通路和雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路在损伤修复和器官纤维化方面发挥着重要的协同作用。
分泌型TGF-β前体二聚体在构象变化、蛋白酶切或者整合素作用下形成具有生物活性TGF-β二聚体,激活TGF-β1以及TGF-β2受体,并活化下游的受体型Smad、共同通路型Smad,形成1个共同的复合物,以此将信号传至细胞核内,激活编码ECM、基质金属蛋白酶、结缔组织生长因子(connective tissue growth factor,CTGF)和snail基因的表达,最终促进EMT的形成[3]。有研究发现,多种干扰TGF-β/Smad信号通路方法,如直接抑制细胞因子、敲除Smad3基因等方法,在肺纤维化的临床模型中均有保护作用[4]。
mTOR是一种进化上十分保守的丝氨酸/苏氨酸蛋白激酶,属于磷脂酰激酶相关激酶(phosphatidyl kinase-related kinase,PIKK)超家族[5]。mTOR的经典道路活化起始于生长因子、营养因子等信号对受体酪氨酸激酶(receptor ftyrosine kinases,RTKs)的活化,主要通过磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/Akt/mTOR和/或细胞外信号调节激酶(extracelluar signal-regulated kinases,ERK)/mTOR两条信号通路,进而影响mTOR的活化。此外,ERK也可直接或间接调节mTOR。mTOR进而磷酸化下游的4E结合蛋白(4E binding protein,4E-BP)和p70核糖体S6蛋白激酶(P70 ribosomal S6 protein kinase,p70S6k)[6]。
近年研究发现,mTORC1/4E-BP信号是TGF-β1促进人胶原合成的关键步骤,并且排除了mTOR通过p70S6K介导TGF-β1的纤维生成作用[7]。典型的Smad3和mTORC1可起到协同作用。经典的TGF-β/Smad3信号道路通过传递信息影响相关基因转录,并且影响mTORC1/4E-BP信号通路的激活;反过来,mTORC1/4E-BP信号通路直接或间接(通过影响相关基因转录)影响TGF-β诱导胶原的表达[7]。有研究显示,mTOR/4E-BP信号可能通过调节mRNA翻译,多步骤地调节TGF-β1促进 EMT反应[6]。
综上,在IPF中,许多信号通路导致组织修复过度、肺组织结构紊乱,其中TGF-β/Smad信号通路是一条重要的影响通路。mTORC1/4E-BP通过调节mRNA转录,进而影响TGF-β/Smad轴,与其发挥协同作用。
白细胞介素-18白细胞介素-18(interleukin-18,IL-18)是一种促炎细胞因子,属于IL-1基因家族。IL-18的增加与一些疾病有相关性,如急性呼吸窘迫综合征、心脏纤维化、肾缺血再灌注损伤及肾纤维化[7-9]。有研究表明,IL-18可能通过以锌指转录因子SNAIL-1依赖的方式促进EMT,增加博来霉素诱导的IPF发展的可能[10]。
血管内皮生长因子近年来,人们对血管内皮生长因子(vascular endothelial growth factor,VEGF)在IPF发病机制中的作用越来越感兴趣。VEGF是目前最受关注、最具代表性、活性最强的血管生成促进因子之一。VEGF家族包括VEGF-A、VEGF-B、VEGF-C、VEGF-D、VEGF-E和胎盘生长因子,其中,VEGF-A主要来自于气道上皮细胞,对正常肺泡分化、肺成熟和血管床发育至关重要,但其确切机制尚未明确[11]。
在许多报道中,VEGF-A作为IPF保护和促进因素存在着矛盾。最近有研究表明,VEGF-A可分为VEGF-AxxxA亚型和VEGF-AxxxB亚型[11]。一项关于小鼠纤维化的临床研究发现,VEGFAxxxA家族抑制VEGFAxxxB家族;低氧环境广泛存在于IPF中,而非正常细胞中。低氧诱导因子-1(hypoxia-inducible factor,HIF-1)直接调节VEGF的转录,在低氧条件下,VEGF-AxxxA亚型特异性上调,增加了VEGF-A基因优先剪接到VEGF-AxxxA亚型的可能性[12]。TGF-β是一种高效的促纤维化细胞因子,可与低氧协同作用,在多个细胞系中刺激VEGF基因表达[13]。与正常肺相比,IPF肺组织中VEGF-A165B明显上调,推测这可能是一种补偿机制,但仍不能避免患者肺发生纤维化[12]。
总的来说,VEGF-AxxxA与血VEGF-AxxxB的比例变化可以被视为疾病的起因,也可以被视为Ⅱ型肺泡上皮细胞和其他细胞的保护性反应,是一种维持体内平衡的调节机制。
血小板衍生因子血小板衍生因子(platelet-derived factor,PDGF)可分为PDGFⅠ和PDGFⅡ,两者均由两条高度同源的A和B组成,这使PDGF具有PDGF-AA、PDGF-AB和PDGF-BB 3种形式的二聚体结构[14]。PDGF在肺纤维化的整个发展过程中发挥作用。研究发现,铝液中PDGF-AB和PDGF-BB水平不仅在晚期升高,而且在早期也升高[14]。PDGF对纤维细胞是一种重要的化学吸引剂,在肺纤维化早期促进纤维细胞向受损肺募集[15]。血小板衍生生长因子受体(platelet-derived growth factor receptors,PDGFR)是一组异质性受体,分为PDGFR-α和PDGFR-β。PDGF与PDGFR-α和PDGFR-β结合后促进受体二聚化,激活PDGF信号。PDGFR-α和PDGFR-β在肺纤维化发病过程中起着不同的作用[16]。Prasad等[17]研究发现,在纤维化肺中,PDGFR-β依赖性通路比PDGFR-α通路更能促进肺纤维化的进展。因此,抗PDGFR-β抗体对IPF患者是一种无肺毒性的、有效的治疗方法。
成纤维生长因子成纤维生长因子(fibroblast growth factor,FGF)是进化上保守的多肽,对关键的发育途径(如形态发生、代谢控制和损伤后再生)起到了举足轻重的影响[18],被认为与肺纤维化的发病机制有关。最近一项研究表明,在IPF肺中,成纤维细胞生长因子受体(fibroblast growth factor,FGFR)1和FGF1的表达发生了改变,FGF信号对肺纤维化中的成纤维细胞迁移至关重要[19]。在人类IPF活检中发现,成纤维细胞病灶的肌成纤维细胞中存在FGF1~4的表达[19]。FGF3、FGF4被证实对早期肺泡发育至关重要[20],FGF3、FGF4联合缺乏的小鼠会有肺弹性纤维沉积[21-22]。FGF也可对IPF起到保护作用。有研究发现,在整个切片的肺纤维化区域,FGFR10、FGFR1和FGFR2的表达水平与肺总量呈负相关[23]。FGF7/FGF10/FGFR2信号通路可能不参与肺重构的初始触发[23]。FGF10的过度表达可保护小鼠免受博莱霉素诱导的肺纤维化也证实了这一假设。但是不能排除这种信号通路可能确实是损伤的介质,甚至是无效的抗纤维化介质或逆转重塑介质[24]。有研究发现,IPF中FGF9和FGF18增加,刺激成纤维细胞迁移,减少成纤维细胞凋亡[25-26]。
间充质细胞在肺纤维化中的研究进展
EMTEMT是胚胎发育过程中一个高度协调的过程,伴随着上皮细胞标记物的消失和间质细胞特征的出现。EMT在IPF的作用充满争议。体外研究表明,EMT发生在经TGF治疗过的Ⅱ型肺上皮细胞[27]和肺间皮细胞[28]。有研究显示,肺泡Ⅱ型和基底上皮细胞可能产生胶原、波形蛋白和S100A4表达的成纤维细胞,目前没有确凿证据表明这些细胞能产生平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)表达的肌成纤维细胞[28]。肺间皮细胞是肺胸膜内的细胞,而肺重塑首先出现肺胸膜处似乎也证明了这点。有研究表明,肺间皮细胞可能通过其增殖和间质转化,在不同的纤维化小鼠模型和IPF中促进胸膜下纤维化反应,从而生成成纤维细胞和肌成纤维细胞[29]。
总之,上皮细胞或内皮细胞通过细胞EMT转化为肌成纤维细胞、成纤维细胞。目前关于两者转变机制的研究仍较为欠缺,有待进一步探究。
成纤维细胞固有成纤维细胞是组织间充质细胞,位于邻近肺泡上皮细胞未受损肺间质。成纤维细胞主要通过活化成肌成纤维细胞、ECM沉积等形式参与肺纤维化[30]。在IPF患者中,原发性人肺成纤维细胞(human lung fibroblast,HLF)端粒缩短,细胞凋亡可能会导致胶原、纤维连接蛋白和其他ECM物质过度积累,导致肺结构发生变化;在IPF患者HLF中观察到一种促纤维衰老相关的分泌表型,特别是在富含TGF-β的小环境中,它可能也与IPF中观察到的纤维化过程有关[31]。
脂质成纤维细胞脂质成纤维细胞是一种富含中性脂质的常驻肺成纤维细胞,对Ⅱ型肺泡上皮细胞的稳态具有重要的意义。lvarez等[31]研究发现,PDGFRα表达的间质成纤维细胞或脂肪成纤维细胞对肺损伤有反应并分化为肌成纤维细胞,而肺重构需要肌成纤维细胞转化为脂肪成纤维细胞。
肌成纤维细胞肌成纤维细胞是肺纤维化的关键效应细胞,是ECM主要的建设者和重塑者,其细胞具有成纤维细胞(显著的内质网)的分泌特点和类似平滑肌(微丝束)的收缩特征。近年来,在IPF动物模型的肺组织中发现了大量肌成纤维细胞,根据其细胞学特征分泌α-SMA可与肌肉中其他蛋白进行区分。一直以来,肺肌成纤维细胞被认为来自于肺周细胞、成纤维细胞、上皮细胞、内皮细胞和骨髓源性干细胞。但近来研究发现,肺肌成纤维细胞群起源于外周细胞、间质成纤维细胞和间质干细胞,但没有来自于上皮细胞、内皮细胞或造血细胞[32-33]。
巨噬细胞巨噬细胞表现出多种功能,一直以来被认为是纤维化过程的中心贡献者。有大量证据支持这样一个观念:巨噬细胞产生多种细胞因子(包括IL-1、PDGF、TGFβ等)调控纤维化反应[34]。根据不同的激活状态,巨噬细胞可分为M1型和M2型2种亚型,M1型称作经典活化型,M2型称作选择活化型。一般而言,M1巨噬细胞通常与炎症相关,可能在纤维化发展过程中起保护作用,而M2巨噬细胞参与基质沉积和组织重塑,促进不受控制的修复[35]。有研究者发现,肺纤维化病灶中的巨噬细胞主要以M2为主。间充质干细胞(mesenchyma stem cell,MSC)是肌成纤维细胞的主要来源[36]。M2巨噬细胞能够激活Wnt/β-连环蛋白信号促进MSC的肌成纤维细胞分化。巨噬细胞另一个参与纤维化的机制可能是诱导TGFβ的转录激活。国外动物实验表明,肺泡巨噬细胞在人和小鼠中都能够产生TGFβ,cAMP效应元件介导的溶解素基序表达细胞中TGFβ1表达的去除阻止了胶原的积累[37]。
ECM
在特发性肺纤维化中,存在着胶原合成与降解间动态失衡,胶原合成增加及除去减少,导致了肺间质中ECM过度沉积。肺内ECM最多的成分是胶原蛋白,以Ⅰ型和Ⅲ型胶原为主。在IPF纤维化早期以Ⅲ型胶原增加为主;晚期以Ⅰ型胶原增加为主,Ⅲ型胶原减少。这种变化是由于胶原酶活性下降、酶抑制物的活性增高,致使胶原分解速率下降。Ⅲ型前胶原氨基端肽(procollagen Ⅲ N-term,P-Ⅲ-NP)是Ⅲ型前胶原在细胞沉积前,在内切酶的作用下所产生的氨基端多肽。在此过程中,Ⅲ前胶原与P-Ⅲ-NP等分子进入血,可以很好地反映胶原合成情况[38]。层黏连蛋白(laminin,LN)存在于基底膜的透明层中,是基底膜特有的一种大分子非胶原糖蛋白,影响细胞的黏附、生长和分化等。 LN在IPF晚期明显增多,吸引黏附成纤维细胞、炎性细胞积聚于基底膜,损伤肺组织,刺激成纤维细胞和上皮细胞分泌胶原,导致肺纤维化[39]。透明质酸(hyaluronic acid,HA)是肺内的一种大分子的糖胺聚糖,其中成纤维细胞在氧自由基等致病因子的作用下使其大量合成,在肺纤维化中有着一定的作用[39-40]。Su等[40]研究发现,IPF患者血清中LN、P-Ⅲ-NP和HA显著升高,可以反映IPF进展,作为其诊断的无创性指标。
UPR对IPF的研究进展
任何干扰蛋白质加工的情况都可能导致内质网中错误折叠蛋白质的积累,称作内质网应激(endoplasmic reticulum stress,ERS)。为了补偿ERS中的细胞损伤和蛋白质折叠紊乱,诱导了UPR信号传导。UPR、转录激活因子(activating transcription factor 4,ATF4)、剪切型X-box结合蛋白1(spliced X-box binding protein 1,XBP-1S)和ATF6α转录因子的3个分支被激活以抑制蛋白质翻译、激活ERS伴侣转录和ERS激活降解。如果内质网功能障碍严重或延长,可通过几种UPR依赖的下游机制诱导上皮凋亡,包括c/ebp同源蛋白(c/ebp homologus protein,CHOP)的诱导、内质网结合半胱天冬酶的激活或c-jun NH2末端激酶的激活。其中CHOP诱导细胞凋亡的研究最为充分,UPR所有3个臂都可以参与CHOP诱导。CHOP通过诱导靶基因bcl2、bim和chac-112,13,14的调节而导致纤维化重塑[41]。有研究者通过研究选择性药物抑制UPR的IRE1α臂,抑制XBP1剪接,防止TGF-β1过度刺激的IPF肺成纤维细胞的促纤维化反应。结果表明,UPR基因表达和Xbp1基因加工和剪接改变可能是IPF纤维化的驱动力[42-43]。局部肺泡缺氧或至少在肺泡上皮细胞中稳定HIF-1α可触发ERS和相关细胞损伤,从而促进肺纤维化的发生[43]。尽管IPF肺中HIF-1α稳定的原因不明确,但针对HIF/UPR/CHOP途径的治疗策略可能是限制肺纤维化发展的一个新的治疗手段。
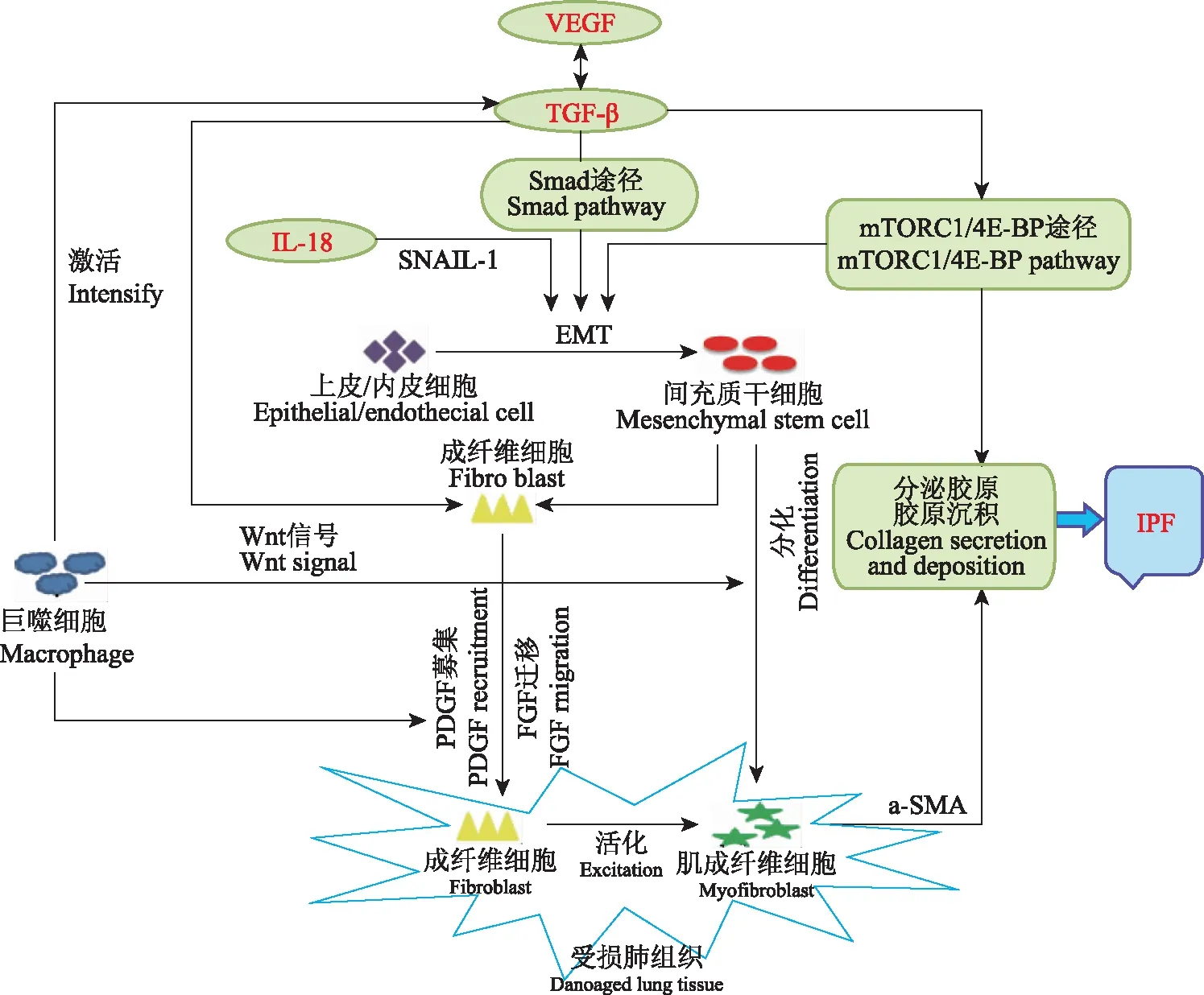
VEGF:血管内皮生长因子;TGF-β:转化生长因子-β;IL-18:白细胞介素-18;PDGF:血小板衍生因子;FGF:成纤维生长因子;EMT:上皮-间充质转化;IPF:特发性肺纤维化;α-SMA:平滑肌肌动蛋白
VEGF:vascular endothelial growth factor;TGF-β:transforming growth factor;IL-18:Interleukin-8;PDGF:platelet-derived growth factor;FGF:fibroblast growth factor;EMT:epithelial mesenchymal transition;IPF:idiopathic pulmonary fibrosis;α-SMA:α-smooth muscle actin
图1特发性肺纤维化与细胞微环境中细胞调节因子、间充质细胞、细胞外基质之间的关系图
Fig1Relationships of idiopathic pulmonary fibrosis with cytokines,mesenchymal cells,and extracellular matrix in cellular microenvironments
综上,IPF是一种由多因素导致的肺间质纤维化疾病。一方面反复损伤的肺泡上皮细胞可分泌TGF-β、VEGF等细胞因子,诱导成纤维细胞及肌成纤维细胞灶的形成;另一方面发生EMT,通过以TGF-β为主多条复杂途径调控EMT的发生发展。
猜你喜欢
中老年保健(2022年2期)2022-11-25
食品与发酵工业(2022年17期)2022-09-17
皮肤性病诊疗学杂志(2022年3期)2022-08-01
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年4期)2021-07-23
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
世界最新医学信息文摘(2021年6期)2021-01-06
体育科研(2013年2期)2013-05-31
