
医院胶囊制剂微生物限度检查方法的建立及结果分析
2020-07-06 06:28:26李亚楠李甜甜杨莉娜杨晓东
中国医院用药评价与分析 2020年2期
李亚楠,李甜甜,沙 淼,杨莉娜,杨晓东
(西安市食品药品检验所微生物室,陕西 西安 710054)
微生物限度检查方法是指非无菌制剂及其原料、辅料受到微生物污染程度的检查方法,其中包括微生物计数法和控制菌检查法。该方法可用于判断非无菌制剂及其原料、辅料等是否符合《中华人民共和国药典》的规定,也可用于指导生产过程中间产品微生物质量监控[1]。检查项目包括需氧菌总数测定、霉菌和酵母菌总数测定以及控制菌检查。本实验研究所用的3种医院胶囊制剂(黄参胰生胶囊、祛风宁胶囊和松萜灵胶囊)为含多种中药成分的复方制剂,处方配伍较复杂,其中所含的黄参、黄芪、川楝子及丹参等均具有较强的抑菌能力[2-4]。含抑菌成分的制剂在进行微生物限度检查时,需消除制剂的抑菌活性后再根据《中华人民共和国药典》规定的方法进行检查[5-7]。对3种医院胶囊制剂按照《中华人民共和国药典:四部》(2015年版)“非无菌产品微生物限度检查”中微生物计数法(通则1105)和控制菌检查(通则1106)分别进行方法验证,发现胶囊制剂对于细菌的抑菌性极高,但在拆分胶囊壳分别对胶囊剂内容物和胶囊壳进行试验后,发现药物本身通过常规方法即可完成微生物限度检查。目前,对胶囊制剂的微生物限度检查普遍集中于如何消除药物的抑菌性,但鲜有考虑该抑菌性是否由于胶囊壳自身原因引起。本研究旨在提示进行胶囊制剂微生物限度检查采用常规方法难以消除药物抑菌性时,建议关注胶囊壳是否在其中起到抑菌作用,从而为微生物限度检查法提供新思路。
1 材料与方法
1.1 材料
1.1.1 仪器:LRH-250型需氧菌及控制菌培养箱(上海一恒科技有限公司);MJ-Ⅱ型霉菌和酵母菌培养箱(上海一恒科技有限公司);SX-500型压力蒸汽灭菌器(日本TOMY公司)。
1.1.2 药品及培养基:3种医院胶囊制剂(黄参胰生胶囊、祛风宁胶囊和松萜灵胶囊)均由西安市某医院提供。胰酪大豆胨琼脂培养基(tryptose agar,TSA)、沙氏葡萄糖琼脂培养基(Sabouraud’s dextrose agar,SDA)、胰酪大豆胨液体培养基(trypticase soy broth,TSB)、麦康凯液体培养基、肠道菌增菌液体培养基及沙氏葡萄糖液体培养基(Sabouraud’s dextrose broth,SDB)由北京奥博星生物技术有限责任公司生产;麦康凯琼脂培养基、木糖赖氨酸脱氧胆盐琼脂培养基由北京三药科技开发公司生产;紫红胆盐葡萄糖琼脂培养基、三糖铁琼脂培养基由北京陆桥技术股份有限公司生产;RV沙门菌增菌液体培养基由广东环凯微生物科技有限公司生产。
1.1.3 菌株:金黄色葡萄球菌[CMCC(B)26003]、大肠埃希菌[CMCC(B)44102]、铜绿假单胞菌[CMCC(B)10104]、枯草芽孢杆菌[CMCC(B)63501]、乙型副伤寒沙门菌[CMCC(B)50094]、白色念珠菌[CMCC(F)98001]和黑曲霉[CMCCF(F)98003]均购自中国食品药品检定研究院,且为第2代。
1.2 方法
1.2.1 菌液的制备:取金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌和白色念珠菌新鲜培养物,分别用0.9%无菌氯化钠溶液10倍递增稀释,制成含菌数<10 000 CFU/ml的菌悬液,备用;取黑曲霉新鲜培养物,用含0.05%(ml/ml)聚山梨酯80的0.9%无菌氯化钠溶液3~5 ml,将孢子洗脱,然后采用适宜的方法吸出孢子悬液至无菌试管内,用含0.05%(ml/ml)聚山梨酯80的0.9%无菌氯化钠溶液制成含孢子数<10 000 CFU/ml的孢子悬液,备用;取大肠埃希菌、铜绿假单胞菌和沙门菌新鲜培养物,分别用0.9%无菌氯化钠溶液10倍递增稀释,制成含菌数<100 CFU/ml的菌悬液,备用。
1.2.2 供试液的制备:称取供试品10 g,加入pH为7.0的无菌氯化钠-蛋白胨缓冲液(耐胆盐革兰阴性菌检查用TSB作为稀释剂)至100 ml,混匀,制成1∶10的供试液。取1∶10供试液50 ml,加入pH为7.0的无菌氯化钠-蛋白胨缓冲液至100 ml,混匀,制成1∶20的供试液。取1∶10供试液20 ml,加入pH为7.0的无菌氯化钠-蛋白胨缓冲液至100 ml,混匀,制成1∶50的供试液。取1∶10供试液10 ml,加入pH为7.0的无菌氯化钠-蛋白胨缓冲液至100 ml,混匀,制成1∶100的供试液。
1.2.3 需氧菌总数测定方法适用性试验:(1)试验组。取上述1∶10(或1∶20)的供试液分装于5个无菌试管中,每管9.9 ml,分别加入金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、白色念珠菌和黑曲霉菌悬液(<10 000 CFU/ml)0.1 ml,混匀后分别注入培养皿,每皿1 ml,33 ℃下培养5 d,测定试验组菌数。(2)供试品对照组。取上述1∶10(或1∶20)的供试液,以菌液稀释液(0.9%无菌氯化钠溶液)代替菌液同试验组操作,测定供试品本底菌数。(3)菌液组。取pH为7.0的无菌氯化钠-蛋白胨缓冲液替代供试液,按试验组操作加入菌液,测定各试验菌菌数。
1.2.4 霉菌和酵母菌总数测定方法适用性试验:(1)试验组。取上述1∶10的供试液分装于2个无菌试管中,每管9.9 ml,分别加入白色念珠菌、黑曲霉菌悬液(<10 000 CFU/ml)0.1 ml,混匀后分别注入培养皿,每皿1 ml,23 ℃下培养7 d,测定试验组菌数。(2)供试品对照组。取上述1∶10的供试液,以菌液稀释液(0.9%无菌氯化钠溶液)代替菌液同试验组操作,测定供试品本底菌数。(3)菌液组。取pH为7.0的无菌氯化钠-蛋白胨缓冲液替代供试液,按试验组操作加入菌液,测定各试验菌菌数。(4)需氧菌、霉菌和酵母菌总数按《中华人民共和国药典:四部》(2015年版)“微生物回收”规定的方法进行微生物计数,若(试验组菌落数-供试品对照组菌落数)/菌液组菌落数的比值在0.5~2.0范围内,认为该方法可用于该样品的菌落计数。
1.2.5 大肠埃希菌检查方法适用性试验:(1)试验组。取TSB 1份(100 ml),加入1∶10的供试液10 ml和<100 CFU的大肠埃希菌。(2)阳性对照组。取TSB 1份(100 ml),加入与试验组相同的菌液。(3)阴性对照组。取TSB 1份(100 ml),加入稀释液10 ml。(4)供试品对照组:取TSB 1份(100 ml),加入1∶10的供试液10 ml。按《中华人民共和国药典:四部》(2015年版)通则1106“大肠埃希菌”项下方法进行试验。
1.2.6 沙门菌检查方法适用性试验:(1)试验组。取TSB 1份(200 ml),加入10 g供试品和<100 CFU的乙型副伤寒沙门菌。(2)阳性对照组。取TSB 1份(200 ml),加入与试验组相同的菌液。(3)阴性对照组。取TSB 1份(200 ml),加入稀释液10 ml。(4)供试品对照组。取TSB 1份(200 ml),加入10 g供试品。按《中华人民共和国药典:四部》(2015年版)通则1106“沙门菌”项下方法进行试验。
1.2.7 耐胆盐革兰阴性菌检查方法适用性试验:(1)试验组。取肠道菌增菌液体培养基6管(每管10 ml),分为两组各3管,第1组分别加入1∶10、1∶100和1∶1 000的供试液各1 ml,同时每管加入<100 CFU的大肠埃希菌;第2组的方法同第1组,分别加入<100 CFU的铜绿假单胞菌。(2)阳性对照组。取肠道菌增菌液体培养基2管(每管10 ml),分别加入与试验组相同的大肠埃希菌和铜绿假单胞菌。(3)阴性对照组。取肠道菌增菌液体培养基1管(10 ml),加入稀释液1 ml。(4)供试品对照组。取肠道菌增菌液体培养基3管(每管10 ml),分别加入1∶10、1∶100和1∶1 000的供试液各1 ml。按《中华人民共和国药典:四部》(2015年版)通则1106“耐胆盐革兰阴性菌”项下方法进行试验。
2 结果
2.1 1∶10供试液平皿法需氧菌总数及霉菌、酵母菌总数测定结果
1∶10供试液平皿法测定3种医院胶囊制剂对铜绿假单胞菌、金黄色葡萄球菌、枯草芽孢杆菌、白色念珠菌和黑曲霉的回收率见表1。由表1可见,上述供试品对白色念珠菌和黑曲霉无显著抑制作用,回收率在0.5~2.0范围内,符合《中华人民共和国药典》关于“微生物回收”的要求;但铜绿假单胞菌、金黄色葡萄球菌和枯草芽孢杆菌的回收率均为0,说明1∶10供试液对细菌的抑菌性极强,需对供试液进行稀释。
2.2 1∶20、1∶50和1∶100供试液平皿法对3种细菌的回收率测定结果
1∶20、1∶50和1∶100供试液平皿法测定3种医院胶囊制剂对铜绿假单胞菌、金黄色葡萄球菌和枯草芽孢杆菌的回收率见表2。由表2可见,将供试液稀释至1∶100时,上述3种细菌回收率仍为0,未能消除供试液的抑菌性。
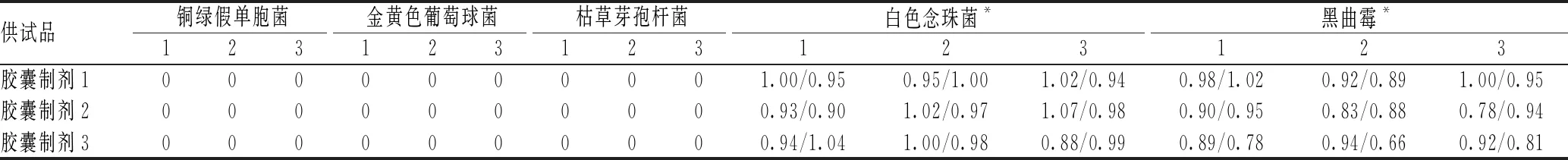
表1 1∶10供试液平皿法回收率测定结果(%)
注:“*”表示回收率分别为TSA和SDA培养基上的2组结果
Note: “*” indicates that the recoveries are the results of two groups on TSA and SDA media
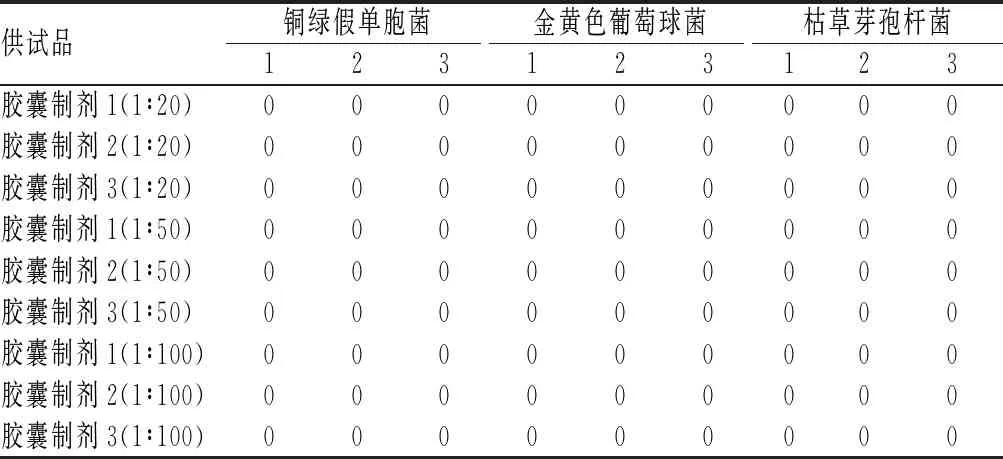
表2 1∶20、1∶50和1∶100供试液平皿法对3种细菌的回收率测定结果(%)
2.3 控制菌检查方法适用性试验结果
分别取1∶10、1∶100和1∶1 000供试液1 ml进行耐胆盐革兰阴性菌检查;取1∶10供试液10 ml进行大肠埃希菌检查;取供试品10 g进行沙门菌检查,结果见表3。由表3可见,3种医院制剂1∶10、1∶100供试液取1 ml,加入10 ml肠道菌增菌液体培养基体积未能使革兰阴性菌生长,1∶1 000供试液取1 ml时方可使革兰阴性菌生长;在将肠道菌增菌液体培养基体积扩大至50 ml时,1∶100供试液1 ml试验组可检出耐胆盐革兰阴性菌;分别将TSB的体积扩大至1 500、3 000 ml,仍未检出大肠埃希菌和沙门菌,说明培养基稀释法未能消除上述3种制剂的抑菌性。
2.4 胶囊制剂内容物、胶囊壳需氧菌总数及霉菌和酵母菌总数测定结果
胶囊制剂内容物、胶囊壳平皿法回收率测定结果见表4。由表4可见,1∶20供试品内容物对铜绿假单胞菌、金黄色葡萄球菌、枯草芽孢杆菌、白色念珠菌和黑曲霉无显著抑制作用,回收率在0.5~2.0范围内,符合《中华人民共和国药典》关于“微生物回收”的要求;但1∶20稀释后的胶囊壳对3种细菌的抑菌未消除,细菌回收率为0,随后将胶囊壳浓度稀释至1∶200进行试验,发现胶囊壳的抑菌性仍未消除,3种细菌的回收率为0。

表3 控制菌验证结果
注:“+”表示正常生长;“-”表示未生长;“/”表示已通过数据无需进行试验
Note: “+” indicates normal growth; “-” indicates no growth; “/” indicates the data has passed and no test is required
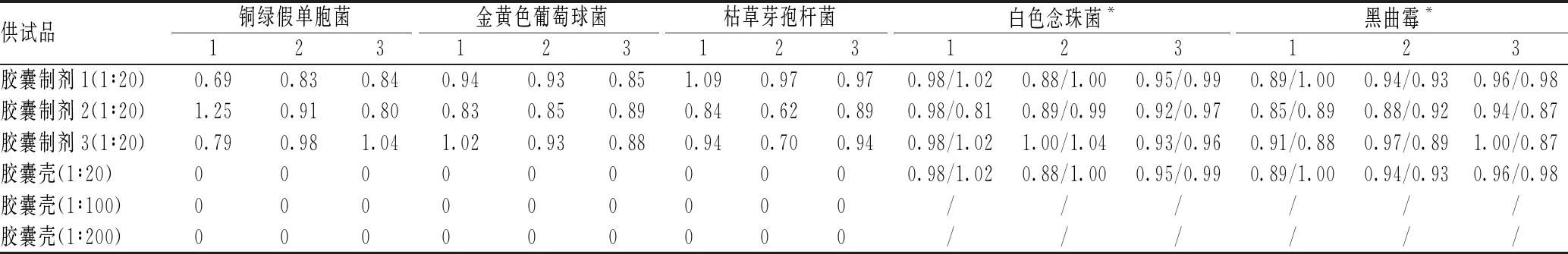
表4 胶囊制剂内容物、胶囊壳平皿法回收率测定结果(%)
注:“*”表示回收率分别为TSA和SDA培养基上的2组结果;“/”表示已通过数据无需进行试验
Note: “*” indicates that the recoveries are the results of two groups on TSA and SDA media; “/” indicates the data has passed and no test is required
2.5 胶囊制剂内容物控制菌检查方法适用性试验结果
胶囊制剂去除胶囊壳后,对内容物进行控制菌检查方法适用性试验,结果显示,耐胆盐革兰阴性菌、大肠埃希菌和沙门菌均可通过《中华人民共和国药典》所规定的常规法检出,见表5。

表5 胶囊制剂内容物控制菌验证结果
注:“+”表示正常生长
Note: “+” indicates normal growth
3 讨论
3种医院胶囊制剂的内容物可按《中华人民共和国药典:四部》(2015年版)通则1105“平皿法”(1∶20供试液,每皿1 ml)进行需氧菌总数、霉菌和酵母菌总数检查;按《中华人民共和国药典:四部》(2015年版)通则1106“常规法”进行控制菌检查。但在不去除胶囊壳的情况下,需氧菌总数检查、控制菌检查均不能通过稀释供试液或扩大培养基体积的方法获得结果。以上结果均表明,胶囊壳中含有显著抑制微生物生长的成分。
胶囊制剂已成为除片剂以外的口服固体制剂的主要剂型之一。明胶胶囊所用明胶是由动物的骨、皮经水解制得,属于蛋白类物质(骨胶或皮胶)[8-11]。明胶胶囊壳作为一种特殊的药用辅料,其质量与药性发挥程度息息相关。劣质胶囊壳不仅会对药品质量产生负面作用,严重时甚至影响人体健康[12]。影响明胶空心胶囊质量的因素繁多,总结归纳大致可分为3类,即重金属含量超标、防腐剂和抑菌剂的超标使用以及兽药、抗菌药物等的残留[13]。重金属含量超标的原因是由于不法生产企业使用皮革废料代替药用明胶,废料中含较多重金属元素(特别是铬离子),而以目前的脱铬技术难以去除铬离子[14-15]。同时,生产企业为了达到胶囊壳微生物指标合格的目的,人为加入过量的防腐剂(如对羟基苯甲酸酯类)和抑菌剂(如环氧乙烷)。此外,明胶的生产材料多为猪、牛和羊等的骨、皮毛,其上残留的兽药、抗菌药物等对明胶空心胶囊的质量优劣形成潜在风险。
目前,关于胶囊壳残留重金属、防腐剂、抑菌剂、兽药及抗菌药物对微生物生长是否产生影响或产生何种影响,并无相关报道。后续实验将对本研究中所使用的胶囊壳进行理化检验,分析其所含成分;同时,针对其成分选取不同种类的中和剂,验证是否可消除胶囊壳的抑菌性,使微生物限度检查方法更加科学、系统和完善。
猜你喜欢
中国测试(2022年4期)2022-05-10 06:29:20
中国土壤与肥料(2021年5期)2021-12-02 01:05:52
疯狂英语·新悦读(2020年7期)2020-07-30 01:32:52
癌变·畸变·突变(2020年1期)2020-02-12 05:18:04
上海蔬菜(2015年2期)2015-12-26 05:03:40
护理研究(2014年26期)2014-08-15 00:50:48
机电信息(2014年35期)2014-02-27 15:54:33
机电信息(2014年20期)2014-02-27 15:53:23
机电信息(2014年5期)2014-02-27 15:51:47
宠物世界·狗迷(2013年7期)2013-04-29 00:44:03
