
稳定表达敲入增强绿色荧光蛋白标记的干扰素 γ受体2基因Ifngr2的黑素瘤细胞系的构建与鉴定
2020-07-02 05:43:12李卫华
中国药理学与毒理学杂志 2020年3期
樊 浩,周 涛,李卫华
(国家生物医学分析中心,北京 100850)
肿瘤的免疫检查点抑制剂治疗以其显著的疗效日益受到人们的重视。目前临床已经批准细胞毒性T淋巴细胞相关蛋白4(cytotoxic T-lymphocyte-associated protein 4,CTLA-4)、程序性死亡受体蛋白 1(programmed cell death protein 1,PD-1)及细胞程序性死亡配体1(programmed cell death ligand-1,PD-L1)的抗体用于治疗晚期黑素瘤、小细胞肺癌和转移性膀胱癌等一系列癌症。然而免疫检查点抑制剂仅对约20%的患者有效,导致治疗失败的原因和机制还未被充分阐明。干扰素γ(interferon-γ,IFN-γ)信号通路在免疫检查点抑制剂治疗中发挥着重要的调控作用。一方面,IFN-γ可以抑制肿瘤细胞的增殖和肿瘤血管的生成发挥直接抗肿瘤作用;另一方面,IFN-γ可以调节肿瘤的转移方式和免疫系统的功能间接抑制肿瘤的生长。近期大量研究表明,IFN-γ信号通路的功能状态决定了免疫检查点治疗的成败[1-3]。
IFN-γ通过与细胞膜上的受体结合,激活下游的信号通路,并调控干扰素诱导基因的表达[4]。IFN-γ受体(IFN-γ receptor,IFN-γR)包含2种不同亚基,分别是IFN-γ受体1(IFN-γ receptor 1,IFNGR1)和IFNGR2[5-7]。研究表明,细胞表面IFNGR1的表达是过量的,发挥着结合IFN-γ的作用[8];而IFNGR2的表达受到严格的调控,其表达量决定了细胞对IFN-γ信号的反应性[9]。明确IFNGR2的表达调控机制有助于阐明IFN-γ信号通路的调控方式和机制,从而为提高肿瘤的免疫治疗效果提供新的策略。
CRISPR-Cas9介导的微同源末端连接敲入技术(microhomology-mediated end-joining)是一种新型的基因编辑技术,利用CRISPR-Cas9造成基因组指定位点的DNA双链断裂,通过细胞内的微同源重组修复机制来实现基因编辑,所需同源序列长度仅约为25 bp,具有载体构建简单,物种应用性广泛,编辑效率高等优势[10]。本研究利用CRISPRCas9介导的微同源末端连接敲入技术将增强绿色荧光蛋白(enhanced green fluorescent protein,EGFP)基因序列敲入到细胞内Ifngr2基因的最后一个外显子末端,使其和Ifngr2基因融合表达,将最新的基因编辑技术应用于IFNGR2表达调控研究,为发现IFNGR2的表达调控因子和靶向肿瘤免疫的药物筛选提供有力工具。
1 材料与方法
1.1 细胞、试剂和仪器
B16黑素瘤细胞(美国模式培养物研究所)。pX330A-1x2,pX330S-2-PITCh 及pCRIS-PITCh(v2)-FBL质粒(美国Addgene质粒共享信息库),BbsI和BsaI核酸内切酶(美国NEB公司),Taq Mix以及T4连接酶(美国Thermo公司),DH5α菌株和6×核酸电泳上样缓冲液(北京博迈德技术有限公司),DNA电泳Marker BM2000和 BM15000,Gen-Green核酸染料(北京博迈德基因技术有限公司),小提质粒试剂盒及中提质粒试剂盒和胶回收试剂盒(美国Promega公司),1640基础培养基和胎牛血清(美国Gibco公司),胰酶和青、链霉素双抗(迈晨科技有限公司),质粒转染试剂LipofectamineTMLTX(美国Invitrogen公司),嘌呤霉素(美国Amresco公司),基因组提取试剂盒(康为世纪公司),质粒测序服务(生工生物工程有限公司)。冷冻离心机5418R以及微量移液器Research®plus系列(美国Eppendorf公司),PCR仪器T100(美国Bio-Rad公司),恒温培养箱3951(美国Thermo公司),超低温冰箱BCD-471WDEA(海尔公司),流式细胞仪FACSCanto Ⅱ(美国BD公司)。
1.2 小引导RNA靶点选择及其寡核苷酸链合成
应用http://chopchop.cbu.uib.no/网站设计Ifngr2小引导RNA(small guide RNA,sgRNA)。本实验设计了2个位点,分别命名为PAM1和PAM2。引物序列如表1所示,引物名称分别为Ifngr2-sgRNA-PAM1和Ifngr2-sgRNA-PAM2,由生工生物工程有限公司合成。设计思路及实验流程如图1所示。
Tab.1 Prime sequences of small guide RNA(sgRNA)

Fig.1 Schematic illustration of CRlSPR/Cas9-mediated Ifngr2-enhanced green fluorescent protein (EGFP)knock-in.A:the target site at the 7thexon close to the stop codon and the donor vector contains a EGFP cDNA for C-terminal fusion;B:general work flow of CRIS-PITCh mediated gene knock-in in B16 cells.
1.3 CRlSPR-Cas9 载体和CRlS-PlTCh(v2)载体构建
构建CRISPR-Cas9载体,首先将sgRNA引物退火形成DNA双链。BbsI酶切pX330A-1x2后进行胶回收。退火引物和酶切载体按比例混合,置缓冲体系,采用T4 DNA连接酶连接。随后,连接产物转化涂板,挑选单克隆后进行菌液鉴定及测序分析。选取序列正确的单克隆扩大培养,提取pX330-1x2-sgRNA质粒。进一步,采用BsaⅠ酶切pX330A-1x2-sgRNA和pX330S-2-PITCh,酶切产物回收纯化后,使用T4 DNA连接酶连接,连接产物转化涂板,然后挑选单克隆进行菌液鉴定及测序比对分析,提取序列正确的阳性克隆质粒,备用。
构建CRIS-PITCh(v2)载体,选定拟敲入EGFP的位置,设计并合成引物(表2),对目的片段和载体分别进行PCR扩增,扩增后片段采用琼脂糖凝胶电泳分离并纯化。将插入片段和载体用重组酶进行同源重组后,重组产物转化涂板,挑取单克隆并进行菌液鉴定以及测序比对分析,提取序列正确的克隆质粒,备用。
1.4 细胞培养和转染
B16细胞使用含10%胎牛血清的1640培养基,在37℃,5%CO2条件下培养。转染前1 d,胰酶消化B16细胞并传代至6孔板,当细胞密度达到60%~80%时,根据LTX转染试剂说明书进行转染操作。设置pmcherry-C1质粒作为阳性对照,将CRISPR-Cas9 载体和 CRIS-PITCh(v2)载体与LTX按比例混匀后静置15 min,然后逐滴加入细胞中。转染6 h后吸除培养液,加入新鲜培养液继续培养过夜,转染24 h后在荧光显微镜下观察阳性对照组荧光情况,以确认转染效果。
1.5 单克隆筛选及EGFP敲入Ifngr2基因细胞系的鉴定
转染72 h后将细胞由6孔板转移至10 cm培养皿,培养基替换为含有嘌呤霉素(2 g·L-1)的培养基,每3 d更换1次培养液,直至培养皿中长出单克隆细胞团。将单克隆细胞团在显微镜下挑至96孔板中继续培养,细胞长满后,转移至6孔板继续培养,待后续鉴定。
首先进行基因组鉴定分析,根据说明书操作,采用康为世纪试剂盒提取基因组DNA,根据插入位点设计不同的不同引物进行鉴定。引物序列见表3。
然后进行蛋白质表达水平分析:①流式细胞分析:将筛选得到的单克隆细胞系胰酶消化后,重悬于流式细胞缓冲液,流式细胞仪检测细胞荧光强度。②荧光显微镜观察:将阳性克隆种在共聚焦专用培养皿中,次日使用荧光显微镜观察EGFP的荧光强度。
1.6 细胞因子及药物处理
将阳性细胞系E10,B4和B16接种至6孔板,分别用 IFN-γ 20 μg·L-1处理 48 h、奥沙利铂2 mmol·L-1处理48 h和TNF-α 20 μg·L-1处理3 h后,与处理相同时间的溶剂(DMSO)对照组同时收样进行后续的流式和PCR实验。

Tab.2 Sequence of primers for target fragments and vector fragments amplification

Tab.3 Sequence of primers for genomic PCR
1.7 实时荧光定量PCR(RT-PCR)分析
细胞用Trizol法提取总RNA,逆转录为cDNA后,对目的基因进行实时定量PCR检测。反应程序如下:95℃ 30 s;95℃ 5 s,60℃ 30 s,40个循环。目的基因相对于内参基因GAPDH的mRNA的表达水平采用 2-ΔCt表示。
1.8 统计学分析
全部实验均经至少3次独立实验。统计学分析均使用GraphPad Prism软件进行,其中组间差异采用t检验进行分析,P<0.05认为差异具有统计学意义。
2 结果
2.1 CRlSPR-Cas9 载体和CRlS-PlTCh(v2)载体的鉴定
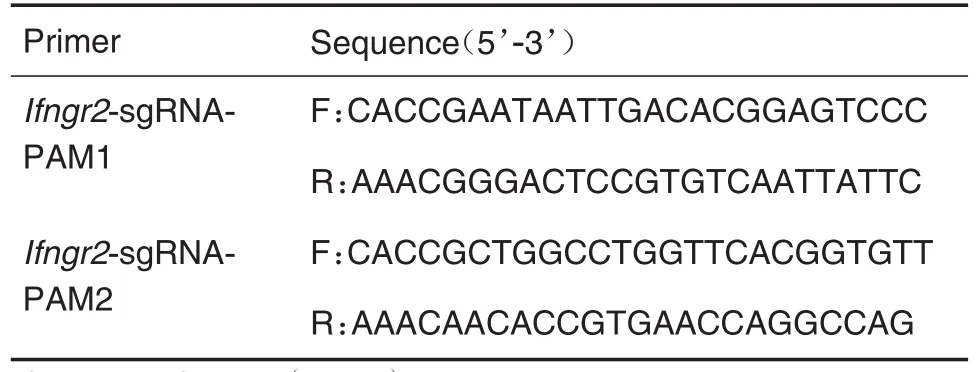
CRISPR-Cas9介导的微同源末端连接基因敲入需要2个载体,载体的主要结构和构建过程如图2A和B所示。其中CRISPR-Cas9载体包含敲入位置的sgRNA和PITCh sgRNA,sgRNA用于制造基因组编辑位置的DNA双链断裂,PITCh sgRNA在供体载体中造成供体片段2端DNA双链断裂。CRIS-PITCh(v2)为供体载体,包含要插入基因组的DNA片段、2A序列分隔的EGFP和嘌呤霉素抗性基因,该片段2端为编辑位置2侧的同源序列,上下游同源臂均约为25 bp。
将含有Ifngr2sgRNA的寡核苷酸链退火后形成DNA双链,然后与BbsI酶切后的pX330A-1x2载体连接,得到载体pX330A-1x2-sgRNA-PAM1和pX330A-1x2-sgRNA-PAM2。用BsaI酶将用于切割供体片段的PITCh sgRNA从载体pX330S-2-PITCh切下,连接入pX330A-1x2-sgRNA载体的BsaI位点,完成CRIPSR-Cas9-PAM1和CRIPSRCas9-PAM2的构建。CRIPSR-Cas9载体凝胶电泳结果如图2C和D所示,载体测序证实CRIPSRCas9-PAM1和CRIPSR-Cas9-PAM2与设计序列一致,表明载体构建成功。
为获得供体载体,设计了分别包含PITCh sgRNA,Ifngr2-PAM1和Ifngr2-PAM2位点上下游序列的引物,对PCRIS-PITCh(v2)-FBL载体中的EGFP-2A-Puro进行PCR扩增,并与扩增后的载体骨架进行重组连接获得CRIS-PITCh(v2)-PAM1和CRIS-PITCh(v2)-PAM2 载体。pCRIS-PITCh(v2)凝胶电泳结果如图2E和F所示,载体测序证实CRIS-PITCh(v2)-PAM1 和 CRIS-PITCh(v2)-PAM2与设计序列一致,表明载体构建成功。
Fig.2 lllustration of vector construction for knock-in and agarose gel electrophoresis of constructed vectors.A:construction of CRISPR-Cas9 vector expressing Cas9 nuclease and two sgRNAs,a target locus-specific sgRNA and a generic PITChsgRNA;B:construction of the CRIS-PITCh(v2)vector harboring the desired microhomologies;C:CRISPR-Cas9-PAM1;D:CRISPR-Cas9-PAM2;E:CRIS-PITCh(v2)-PAM1;F:CRIS-PITCh(v2)-PAM2.M:marker.
2.2 稳定表达敲入EGFP标记的Ifngr2基因B16细胞系的鉴定
将2.1中所述CRIPSR-Cas9和其对应的pCRIS-PITCh(v2)质粒载体同时转入小鼠黑素瘤细胞系B16中。转染后72 h通过嘌呤霉素筛选阳性细胞。设计了3对PCR引物(图3A)对阳性细胞系进行了鉴定。阳性克隆可产生3种PCR产物,分别命名为产物1,2和3,其中产物1上游引物位于基因组上,下游引物位于EGFP上,产物分子质量约500 bp;产物2和3引物位于上下游基因连接处,产物分子质量约1450 bp。克隆E10和B4 PCR结果如图3B和C所示,设计PCR产物分子质量处均出现特异性条带。将产物1纯化后进行测序,使用下游引物测序,比对结果如图3D和E所示,其中测序上游是EGFP序列,中间是连接处的同源臂序列,下游是基因组序列,说明EGFP序列成功插入基因组目标位点。

Fig.3 Verification of lFNGR2-EGFP in B16 cell lines via genomic PCR,flow cytometry and fluorescence microscopy.A:three pairs of primers were designed for genomic knock-in confirmation;B and C:electrophoretic image of genomic PCR(E10 and B4),red arrow pointing to target band;D and E:sequencing results of product 1;F:flow cytometry of the knock-in cells,GFI:green fluorescence intensity;G:knock-in cell imaging using fluorescence microscope.
对基因组PCR鉴定阳性的细胞系进行了流式分析及荧光显微镜下观察。流式结果如图3F所示,以B16细胞为对照组,红色范围内为阳性细胞,横轴表示绿色荧光强度,E10和B4细胞系绿色荧光强度显著高于对照组,阳性率>95%。荧光观察结果如图3G所示,其中B16细胞为对照组,在E10和B4细胞系中可以成功看到绿色荧光,对照组则看不到,说明插入EGFP序列表达成功。
2.3 阳性克隆细胞的lFN-γ反应性检测
为了明确阳性细胞系是否能够响应IFN-γ的刺激,对阳性克隆E10和B4进行IFN-γ(20 μg·L-1)处理48 h并检测下游靶基因Cd274(PDL1)和Psmb9的表达。如图4所示,以B16细胞为对照,纵坐标表示mRNA相对表达量。IFN-γ刺激后,Cd274(PDL1)在E10和B4细胞中显著上调了63和138倍,Psmb9在E10和B4细胞中显著上调了2885和4616倍,上调幅度与B16母细胞相当,说明E10和B4细胞能够响应IFN-γ刺激,表明细胞中IFN-γ信号通路完整。

Fig.4 Real time PCR(RT-PCR)of interferon-γ(lFN-γ)target gene Cd274 and Psmb9 in knock-in cells(E10 and B4)and B16 parental cells.A:analysis of Cd274 in cells with or without IFN-γ 20 μg·L-1treatment;B:analysis of Pmsb9 in cells with or without IFN-γ treatment.±s,n=3.*P<0.05,**P<0.01,compared with corresponding cell control group.
2.4 TNF-α和奥沙利铂上调阳性克隆细胞中lfngr2和EGFP的表达
将细胞克隆E10和B4分别用TNF-α和奥沙利铂刺激后进行流式细胞分析,同时提取RNA进行实时荧光定量PCR分析。流式结果如图5A和E所示,红色为对照组B16,绿色为未加刺激的阳性细胞系,橙色为加药刺激的阳性细胞系,横轴表示绿色荧光强度。阳性克隆E10和B4细胞系加药刺激后的绿色荧光峰值均有右移,说明EGFP表达增加。计算E10和B4主细胞群的平均荧光强度值,TNF-α处理后,E10和B4细胞平均荧光强度由3845和4456上升到5720和5972;奥沙利铂处理后,E10和B4细胞平均荧光强度由3032和2786上升到4285和4179,因此TNF-α和奥沙利铂处理使E10和B4细胞的荧光显著增强(图5B和F)。实时荧光定量PCR分析,TNF-α刺激后,Ifngr2在B16,E10和B4细胞中分别上升11.40,9.31和21.39倍,EGFP在E10和B4细胞中分别上升了2.50和5.37倍;奥沙利铂刺激后,Ifngr2在B16,E10和B4中细胞分别上升10.99,3.54和5.39倍,EGFP在E10和B4细胞中分别上升2.70和2.44倍;IFN-γ处理不能上调Ifngr2和EGFP的表达(图5C,D,G,H,I和J)。以上结果表明,阳性克隆细胞系细胞中绿色荧光的强度可以特异性地反映细胞中Ifngr2的表达水平。
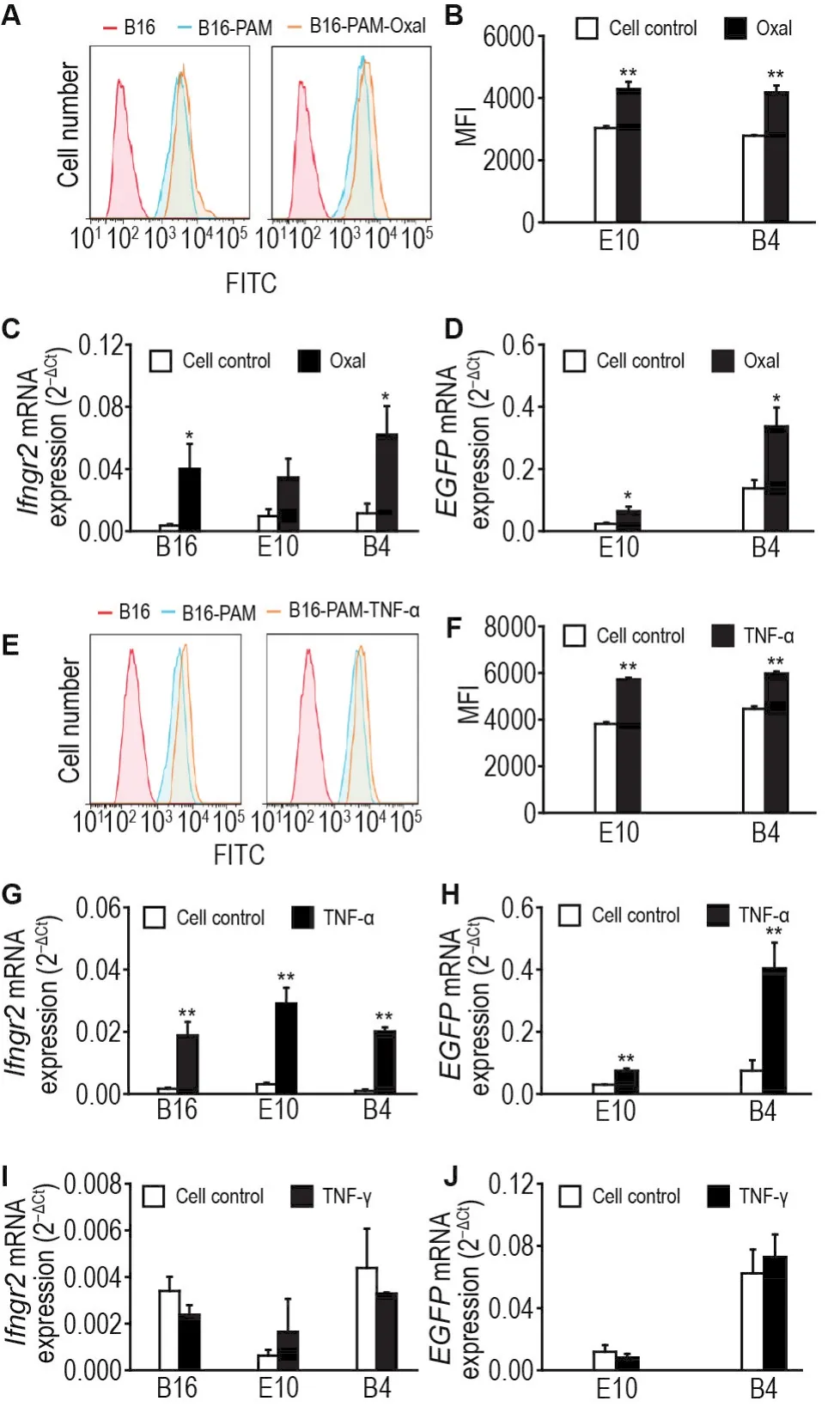
Fig.5 Ifngr2 and EGFP upregulated by tumor necrosis factor-α(TNF-α)and oxaliplatin(Oxal).A and E:flow cytometry analysis of the fluorescence of cells with Oxal 2 mmol·L-1(A),TNF-α 20 μg·L-1(E)or without treatment.B and F:statistical results of flow cytometry observation.MFI:mean fluorescence intensity.±s,n=3.*P<0.05,**P<0.01,compared with cell control group.C,D,G,H,I and J:results of RT-PCR on the indicated genes before and after stimulation.TNF-α and Oxal could upregulate the expression of Ifngr2 and EGFP.But IFN-γ couldn’t.±s,n=3.*P<0.05,**P<0.01,compared with corresponding cell control group.
3 讨论
IFN-γ是由自然杀伤细胞和激活的效应T细胞分泌的一类细胞因子,在机体的免疫过程中发生着十分重要的作用[11-14]。IFN-γ能够促进组织相容性复合体Ⅰ类及Ⅱ类抗原的加工提呈,因此提高机体的免疫监视功能[15]。IFN-γ能够调节Th1细胞的应答,协调固有免疫反应向获得性免疫反应转化[16]。除此之外,IFN-γ还能调节免疫细胞功能,影响免疫反应。
近年来研究证明,IFN-γ信号通路在肿瘤的免疫治疗中具有十分重要的作用。GAO等[1]发现,肿瘤细胞中IFN-γ信号通路的缺失导致了肿瘤细胞对药物的耐受,从而导致抗CTLA4治疗的失败。BENCI等[3]发现,IFN-γ信号通路可以调节肿瘤细胞对免疫检查点药物的敏感性。此外,ZARETSK等[2]也报道了在黑素瘤细胞IFN-γ信号通路相关基因表达的异常,导致抗PD-1治疗失败。IFNGR1在细胞中高表达,IFNGR2的表达受到严格的调控,可以对IFN-γ信号通路实施调控,进而影响肿瘤免疫效果。因此,研究IFNGR2的表达调控具有十分重要的意义。
基因编辑技术是指在细胞基因组靶位点引入核酸序列变化的一类技术,其核心为在打靶位置引入DNA双链断裂,应用细胞内的DNA修复机制,引入对该位置的基因插入、缺失、替换等。同源重组基因编辑技术应用细胞中的同源重组修复机制,可以精确的实现目标基因的替换,已经在人多功能干细胞[17-18]和小鼠[19]等细胞中应用。但是,基因重组其所需同源臂较长(至少200 bp),载体构建困难,编辑效率较低。本研究所采取的CRISPR-Cas9介导的微同源末端连接敲入技术,利用的是细胞内另一种DNA损伤修复机制——微同源末端连接,该机制可以通过~25 bp的同源臂将DNA片段连入双链断裂区域,实现基因敲入,载体构建简单,编辑效率高。本研究采用RNA引导的CRISPR-Cas9实现基因组双链DNA断裂,与锌指核酸酶、转录激活子样效应物核酸酶和第一代RNA引导的CRISPR-Cas核酸酶相比,切割更为高效,可应用的细胞种类更多。CRISPR-Cas9介导的微同源末端连接敲入技术已经在脊椎动物和无脊椎动物动物细胞中得到了成功应用[20-21]。
在抗生素筛选得到了敲入细胞系的候选单克隆后,从基因组水平和蛋白质表达水平筛选EGFP正确敲入的阳性克隆。PCR结果显示,PAM1获得的阳性克隆较多,PAM2阳性克隆较少,提示PAM1和PAM2 2个靶位点的编辑效率存在一定差异。在蛋白质表达水平,通过流式细胞仪和荧光显微镜观察候选克隆的绿色荧光强度。结果发现,即使PCR结果均为阳性的克隆,其荧光强度也有差异,因此保留了荧光强度不同的细胞克隆,其中E10和B4阳性细胞克隆绿色荧光强度中等。不同荧光强度的克隆将可能适用于不同的筛选需求,其绿色荧光强度水平的差异可能与细胞克隆差异和细胞内源表达调控机制相关,具体原因有待进一步研究。
TNF-α和奥沙利铂都是用来治疗肿瘤的药物,已有文献提示它们均可促进抗肿瘤免疫治疗的效果。本研究发现,TNF-α和奥沙利铂处理使E10和B4细胞中的Ifngr2mRNA水平显著上调,提示TNF-α和奥沙利铂促进肿瘤免疫治疗效果可能是通过上调Ifngr2水平进而增强IFN-γ信号实现的。同时,在这2株细胞系中,TNF-α和奥沙利铂处理导致EGFPmRNA水平和绿色荧光强度的显著上调,与Ifngr2的上调一致,表明构建的细胞系中绿色荧光的强度可以反映IFNGR2的表达水平。
作为工具细胞系,需要验证阳性细胞系中绿色荧光强度对于IFNGR2的表达水平的反应程度。同时还需要从其他方面对阳性细胞系进行鉴定,如上下游通路是否正常激活与响应,这仍需要进一步实验验证。
综上所述,本研究采用微同源末端连接基因编辑技术,成功构建了稳定表达敲入EGFP的Ifngr2基因的黑素瘤B16细胞系。该细胞系具有正常的IFN-γ信号通路,且其中绿色荧光强度可反映IFNGR2的表达水平,提示该细胞可用于IFNGR2表达及功能调控研究,为筛选IFNGR2的表达调控因子和潜在的抗肿瘤免疫治疗药物提供有力工具。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
环球时报(2022-09-20)2022-09-20 15:18:57
汉字汉语研究(2021年2期)2021-08-30 08:58:46
今日农业(2020年24期)2020-12-15 16:16:00
兽医导刊(2016年12期)2016-05-17 03:51:50
河北书画研究(2016年3期)2016-04-28 08:55:35
山东医药(2015年14期)2016-01-12 00:39:43
江苏大学学报(医学版)(2015年2期)2015-04-17 06:49:51
中国医药导报(2015年26期)2015-02-28 22:07:44
