
CNTs@Y2O3:Eu3+纳米复合材料的制备及其荧光温敏性能
2020-06-30 06:38王赛楠张春明
功能材料 2020年6期
王赛楠,李 颖,张春明
(上海理工大学 材料与科学工程学院,上海 200093)
0 引 言
近年来,一维稀土纳米无机材料越来越引起人们的兴趣,其在光学、催化、传感及医学等领域具有重要的研究意义和应用价值[1-5]。一维稀土纳米无机材料的制备方法主要包括气相机理生长法[6-7]、溶液生长法[8-10]、自组装生长法[11-13]以及模板法[14-18]等。目前以碳纳米管(CNT)为模板的稀土荧光纳米材料成为研究热点[19-20]。然而,在一定的范围内该类稀土纳米复合材料的荧光性能随温度变化的灵敏性未见公开报道。因此,在本研究工作中,我们利用表面活性剂十二烷基苯磺酸钠(SDBS)为辅助的均相沉淀法,将具有荧光特性的稀土氧化物前驱体与一维结构的碳纳米管(CNT)进行复合,成功制备了CNTs@Y2O3:Eu3+纳米复合材料。研究分析了复合材料的微观形貌结构、热性能、结晶度以及荧光性能,进一步探讨了在400~1 000 ℃温度范围内,煅烧的CNTs@Y2O3:Eu3+复合材料荧光强度的变化,从而反映出该复合材料的荧光温敏特性。
1 实 验
1.1 一维结构CNTs@Y2O3:Eu3+复合材料的制备
1.1.1 制备过程
(1) 稀土氯化物的制备:
将Y2O3·6H2O和Eu2O3·6H2O的氧化物分别与2M HCL反应制得浓度为0.1 %的YCl3溶液和EuCl3水溶液备用。
(2)合成一维结构的CNTs@Y2O3:Eu3+微球:
分别取YCl3溶液 (12 mL 0.1 M)和EuCl3溶液 (0.64 mL 0.1 M) 于100 mL圆底烧瓶中;将尿素(1.5 g)溶于30 mL去离子水,待澄清后加入圆底烧瓶中;取-COOH修饰的CNTs(0.05 g)溶于30 ml去离子水中,加入0.1 g SDBS,超声分散后加入到上述溶液中,将混合溶液再次超声以免发生团聚,90 ℃回流反应4 h;反应结束后抽滤,分别采用去离子水和无水乙醇各清洗3次,并于70 ℃干燥12 h,所得产物收集备用。
(3) 煅烧CNTs@Y2O3:Eu3+:将上述制得的复合材料CNTs@Y2O3:Eu3+置于箱式炉中,空气气氛下分别在不同的温度中煅烧制得样品。
1.1.2 制备机理
Y2O3:Eu3+空心管的均相沉淀法制备示意图如图1,以一维CNTs为模板,采用表面活性剂辅助的均相沉淀法制备均匀的CNTs @ Y2O3:Eu3+复合材料。初始阶段,在H2SO4溶液中经KMnO4化制备表面带有-COOH的碳纳米管模板。之后,通过表面活性剂辅助的均相沉淀的方法制备核壳结构的前驱体Y(OH)CO3:Eu3+,随着反应的进行,壳继续生长。最后,在煅烧过程中,壳层Y(OH)CO3:Eu3+前驱体材料逐渐氧化成一维结构CNTs @ Y2O3:Eu3+。
1.2 样品性能及表征
CNTs@Y2O3:Eu3+纳米棒的形貌分析采用日本日立公司S-4800高分辨场发射扫描电镜;能谱分析用Philips公司TecnalG2 20型透射电子显微镜,在乙醇介质中超声分散30 min后,在铜网碳膜上进行测定分析;通过Pyris I型TGA来研究样品的组分以及热稳定性;荧光光谱采用日本岛津RF-5301型荧光分光光度计测定;红外光谱由美国Nicolet傅立叶红外光谱仪(Nexus)分析测定。
2 结果与讨论
2.1 CNTs @Y2O3:Eu3+与Y2O3:Eu3+的结构及形貌确定
图2分别为未煅烧的CNTs@ Y(OH)CO3:Eu3+(a)前驱体和CNTs@Y2O3:Eu3+(b)复合空心管样品在800 ℃煅烧2 h后的XRD图谱。从图2(a)中可以观察到位于2θ=25°和2θ=45°处出现两个宽带,这表明在煅烧之前,所制备的一维结构的前驱体复合材料是无定形结构。这是由于在未煅烧的复合材料中存在大量无定形结构的一维CNTs,从而影响整个复合材料的晶体结构以及结晶状态。将800 ℃煅烧2 h的CNTs @Y2O3:Eu3+复合空心管样品(图2b)与卡片JCPDS no.65-3178进行对比。XRD曲线在2θ=29°,33.5°,48和57°的衍射峰分别对应于(222),(400),(440)和(622)的晶面,进一步证实了Y2O3:Eu3 +的立方相晶体结构。此外,由图可以观察到Y2O3:Eu3 +样品的衍射峰非常尖锐,这也证实了所制备的产物是纯净的,并且Eu3+均匀地掺入Y2O3主晶格中获得了具有高结晶度的Y2O3:Eu3+。
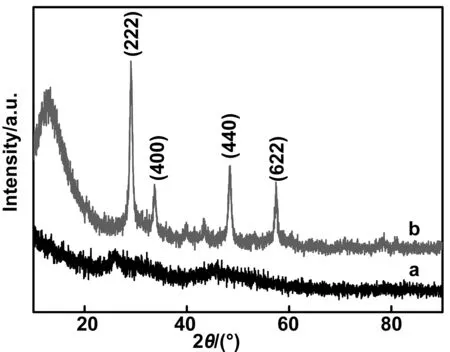
图2 (a)未煅烧的CNTs @ Y(OH)CO3:Eu3 +前驱体和(b)CNTs @Y2O3:Eu3 +复合空心管的XRD图谱Fig 2 XRD patterns of uncalcined CNTs@Y(OH)CO3:Eu3+ precursor and CNTs@Y2O3: Eu3+ composite hollow tube

图3 (a)CNTs(b)前驱体CNTs@Y(OH)CO3:Eu3+(c)800℃煅烧 CNTs@Y2O3:Eu3+复合材料的FT-IR图谱Fig 3 FT-IR spectrum of CNTs, precursor CNTs@Y(OH)CO3:Eu3+ and CNTs@Y2O3:Eu3+ calcined at 800 ℃
通过FT-IR光谱可以分析一维CNTs, CNTs @Y(OH)CO3:Eu3+前驱体和800 ℃煅烧2 h的CNTs@Y2O3:Eu3+复合空心管的表面官能团结构。从图3可以看出,与CNTs的红外光谱(a)相比, CNTs @Y(OH)CO3:Eu3+前驱体(b)中不仅包含了一维CNTs的特征吸收峰,而且位于3 375 cm-1,1513 cm-1,1 403 cm-1,1300-900 cm-1和843 cm-1附近也出现了的吸收峰,分别对应于OH-(ν),CO(ν),CH(δ),CO(νs)和CH(δ)(ν:伸缩振动;νs = 反对称伸缩振动δ:平面弯曲振动),这表明前驱体由CNTs的核结构和Y(OH)CO3:Eu3+壳结构两部分组成。在800 ℃煅烧2 h的CNTs @Y2O3:Eu3+复合空心管的FT-IR光谱中(c),位于563 cm-1的吸收峰归因于Y2O3中Y-O的伸缩振动,证实了Y2O3:Eu3+晶体结构的存在,与XRD图谱的分析结果一致。 另外,从CNTs@Y2O3: Eu3+(图3 c)仍然可以观察到一维CNTs的特征吸附峰,这也说明CNTs模板没有完全去除。
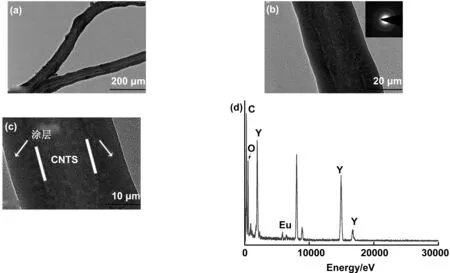
图4 (a)CNTs @ Y(OH)CO3:Eu3+前驱体的TEM图;(b)HRTEM图,插图为选中区域的电子衍射图;(c)展示CNTs涂层的HRTEM图和(d)EDS图谱Fig 4 TEM and HRTEM image of CNTs@Y(OH)CO3:Eu3+ precursor composite; the illustration of b shows the electron diffraction pattern of the selected area, and HRTEM and EDS pattern of CNTs coating

图5 600 ℃煅烧2h的CNTs @ Y2O3:Eu3+复合材料TEM图(a)和HRTEM图(b)及EDS图(e)、800 ℃煅烧2h的CNTs @ Y2O3:Eu3+复合材料TEM图(c)和HRTEM图(d),插图为选中区域的放大图及EDS图(f)Fig 5 TEM image (a), HRTEM image (b) and EDS image (e) of CNTs@Y2O3:Eu3+ composite material calcined at 600 ℃ for 2 h and TEM image (c), HRTEM image (d) and EDS image (e) of CNTs@Y2O3:Eu3+ calcined at 800 ℃ for 2 h, the illustration is an enlarged view of the selected area
为了进一步分析材料的结构,通过TEM图谱进一步探究所制备样品的形貌和结构,如图4所示。图4分别给出了未煅烧的CNTs @ Y(OH)CO3:Eu3+前驱体TEM图(a)、HRTEM图(b)、CNTs涂层的HRTEM图(c)和能谱EDS(d)。由图4 a可以看出,CNTs表面均匀地包覆着一层致密的涂层,图4 b显示出涂层的高分辨率透射电镜(HRTEM)图像,其中,插图为相应选中区域的电子衍射图,衍射图显示是漫射环,表明涂层材料是无定形的。涂层区域如图4 c所示,其厚度约为22 nm。根据EDS能谱分析(图4 d),表明存在C、Eu、Y和O元素,其中(Y、Eu)与O的原子比近似为1∶4,因此可以推断出前驱体材料的化学组成为Y(OH)CO3:Eu3+。
图5显示了CNTs @ Y(OH)CO3:Eu3+前驱体复合材料煅烧后,样品的形态和结构。600 ℃煅烧2 h的CNTs @ Y2O3:Eu3+复合材料TEM图像(图5 a)和HRTEM图(图5 b),从图中可以观察到, CNTs表面存在致密的尺寸较为均匀的涂层,其厚度约为10 nm,样品形状的大小由CNTs模板确定。800 ℃煅烧2h的CNTs @ Y2O3:Eu3+复合材料TEM图像(图5 c)和HRTEM图像(图5 d)显示原先表面致密的涂层呈现出纳米颗粒较为均匀地包覆在CNTs表面的现象,且厚度变为8 nm左右,与未煅烧的CNTs @ Y(OH)CO3:Eu3+前驱体和600 ℃煅烧后的CNTs @ Y2O3:Eu3+复合材料相比,Y2O3:Eu3+壳层的平均厚度逐渐减小,这是由于煅烧过程中CNTs 的分解脱水和壳层中较为松散的前驱体向致密氧化层的转化。从图5 d插图Y2O3:Eu3+壳层局部的HRTEM图像中可以看到明显的晶格条纹,相邻晶格条纹之间的晶面间距约为0.25 nm,对应于立方相Y2O3(JCPDS No.65-3178)的(222)晶面的间距。证实了样品的高结晶度,这与XRD分析结果非常一致。通过EDS图(图5 e和f)对所制备样品的化学组成进行研究。结果显示(Y,Eu)与O的原子比近似为2∶3,因此,可以推断壳层的组成为Y2O3∶Eu3+,EDS分析与XRD结果一致。
2.2 热性能分析
CNTs@Y(OH)CO3:Eu3+前驱体的TG-DTA曲线如图6所示。从曲线中能够观察到两个主要的失重阶段,即79.1 ℃和426.8 ℃质量损失分别为15.31%和23.37%。两次质量损失分别可归因于小分子水的分解和CNTs模板的分解、燃烧(在此过程中,无定形的CNTs@Y(OH)CO3:Eu3+前驱体逐渐向结晶态Y2O3:Eu3+进行转化)。煅烧到1 000 ℃后剩余质量为54.83%,主要包含剩余未煅烧的CNTs和Y2O3:Eu3+壳层。
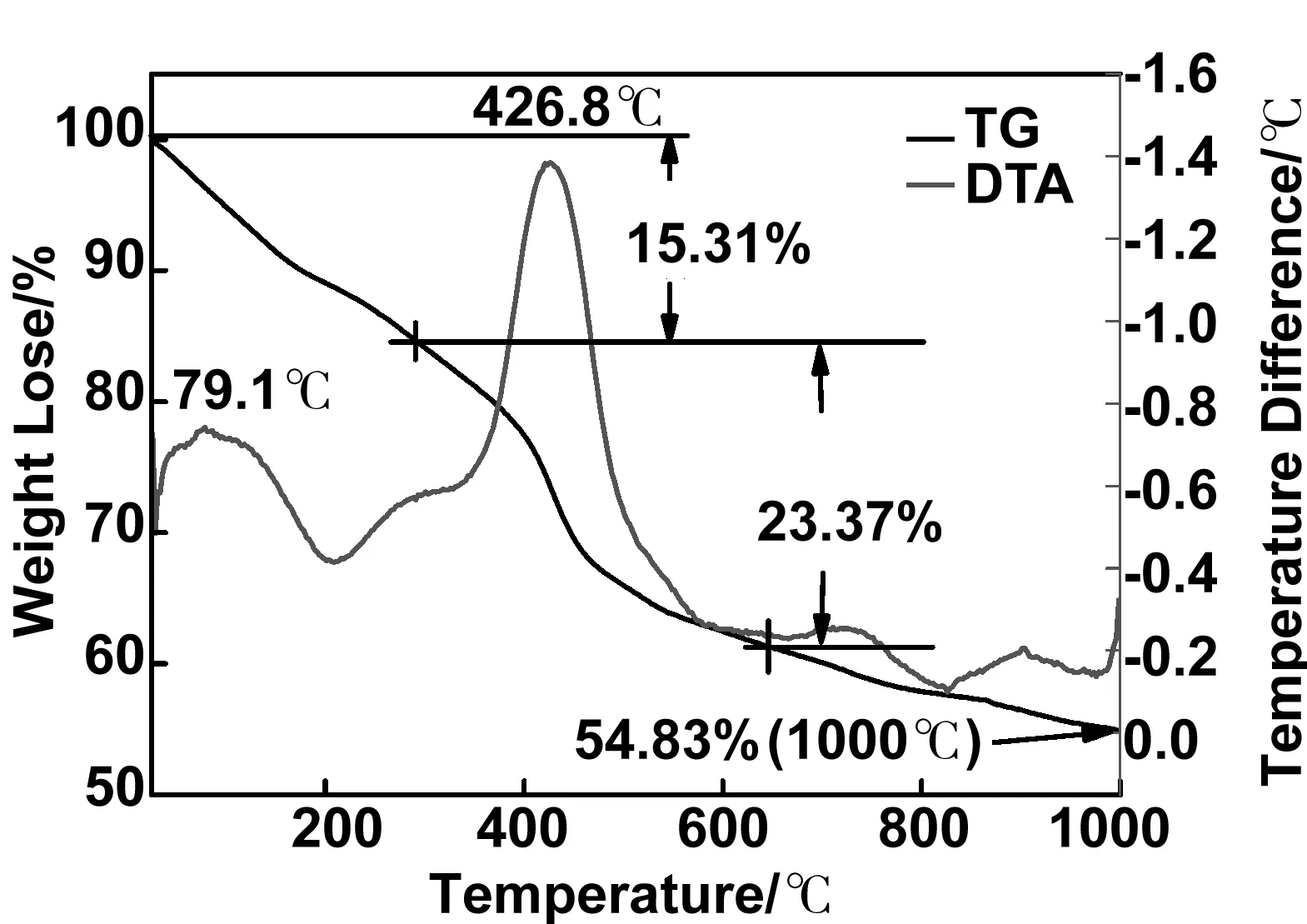
图6 CNTs@Y(OH)CO3:Eu3+前驱体的TG-DTA曲线Fig 6 TG-DTA curve of CNTs@Y(OH)CO3:Eu3+ precursor
图7为CNTs @ Y(OH)CO3:Eu3+前驱体在400~1000 ℃煅烧2h后的激发光谱(a)、发射光谱(b)、三维发射光谱图(c)和在613 nm处的荧光强度变化图(d)。从激发光谱(图7 a)和发射光谱(图7 b)中看到,前驱体复合材料CNTs@Y(OH)CO3:Eu3+在395 nm处具有较强的激发峰,即以395 nm为激发波长可获得材料相应的发射光谱图;其次,900℃发射峰的强度在温度范围内最强,这是由于复合材料的CNTs核煅烧后质量分数逐渐减少,立方晶体结构的Y2O3:Eu3+在高温煅烧后结晶度增强,荧光强度也随之增强。随着煅烧温度的升高,复合材料的荧光强度呈现先增强后减弱再增强的趋势(如图7 c和d所示),呈现出复合材料的荧光强度随温度变化的敏感特性。结合TG-DTA曲线(图6),600 ℃之前,煅烧过程中复合材料中有大量的CNTs模板逐渐分解、气化,这些无定形结构的聚合物破坏了晶体的规整性,复合材料结晶度降低,荧光强度减弱;600 ℃时复合材料中无定形结构减少,材料结晶度提高,荧光强度较强。到900 ℃之前,复合材料有一段质量的损失(CNTs的继续分解),无定形结构进一步减少,结晶度提高,荧光强度较600 ℃煅烧的复合材料更强。到1000 ℃荧光强度稍有下降,这是随着温度的升高产生了荧光淬灭现象。

图7 CNTs@Y(OH)CO3:Eu3+在400~1 000 ℃煅烧后的激发光谱图(a)、发射光谱图(b)、三维发射光谱图(c)和在613 nm处的荧光强度变化图(d)Fig 7 Excitation spectrum (a), emission spectrum (b), three-dimensional emission spectrum (c), and line graph of fluorescence intensity at 613 nm (d) of CNTs@Y(OH)CO3:Eu3+ after calcination at 400-1 000 ℃
3 结 论
采用表面活性剂辅助的均相沉淀法,以一维多壁碳纳米管作为硬模板,成功制备了壳层厚度约为8 nm的具有一维结构的CNTs@Y2O3:Eu3+复合材料。在沉淀过程中,加热,搅拌以及尿素的存在使Y3+和Eu3+在表面活性剂修饰的CNTs模板表面有效和均匀地沉淀。随后在不同的温度下煅烧,获得了结晶度较高的一维结构CNTs @ Y2O3:Eu3+。所制备的复合材料在UV激发下显示出强烈的稀土Eu3+特征荧光。同时,该复合材料在不同煅烧温度下荧光强度随之发生明显变化,具有较强的温度敏感特性,将有望应用于荧光显示器件或生物医学领域等。
猜你喜欢
陶瓷学报(2020年2期)2020-10-27
中国材料进展(2020年4期)2020-05-23
火工品(2019年3期)2019-09-02
天津医科大学学报(2019年3期)2019-08-13
西北药学杂志(2018年5期)2018-09-20
中国资源综合利用(2017年4期)2018-01-22
湖北农业科学(2016年5期)2016-10-19
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
应用化工(2014年11期)2014-08-16
