
碳化铁/氮掺杂碳纳米粒子的制备及其电化学析氢性能
2020-06-30 06:37王国欣
功能材料 2020年6期
杨 森,王 兵,熊 鹰,陈 卓,王国欣
(1. 西南科技大学 材料科学与工程学院,四川 绵阳 621010;2. 西南科技大学 环境友好能源材料国家重点实验室,四川 绵阳 621010)
0 引 言
随着社会经济的快速发展,能源危机和环境污染成为了人类亟待解决的难题,发展环境友好、来源丰富的清洁能源具有极其重要的意义[1]。氢能兼具来源广、燃烧热值高、产物环保、能量密度高和零排放等优点,被认为是一种理想且极具潜力的化石燃料替代品[2]。发展氢能源最重要的就是开发高效低成本制氢技术,现有的制氢技术主要包括燃料制氢、生物制氢、光催化制氢和电解水制氢等,其中,电解水制氢以制备条件温和、生产方式安全、制氢纯度高和对生产设备要求低等优点,受到广泛关注[3-4]。在电解水制氢技术中最为关键的是催化剂材料,常用的铂基贵金属催化剂具有相对较好的催化性能[5-6],但由于其稀缺性和价格昂贵的劣势,限制了其大规模应用[7-8]。因此,设计开发高效、廉价和来源丰富的非贵金属类催化剂材料成为氢能研究领域的重要课题。
在过去十年中,已有大量的非贵金属析氢催化剂被广泛的研究,如碳化物[9]、硫化物[10]、磷化物[11]和金属合金[12]等。其中,过渡金属碳化物由于其具有类似于贵金属铂的d轨道态密度、丰富可调的物相和组成、良好的物理化学稳定性和电子传导性等优点,引起了广泛的关注[13-14]。然而,传统碳化物制备过程中,大多需要高温煅烧,在煅烧过程中往往缺少对碳化物纳米颗粒的保护,这会造成碳化物纳米颗粒的不可控生长、团聚和烧结等现象,从而导致碳化物的活性位点被掩蔽[15]。在众多过渡金属碳化物中,碳化铁(Fe3C)是一种极为重要的过渡金属碳化物,被认为是一种有发展前景的析氢反应催化剂。但Fe3C导电性差,电荷转移电阻大,活性中心密度低的缺点也在一定程度上影响了它的催化性能[16]。为此,研究者们尝试将其与碳材料结合以提高材料的导电性和分散性,从而增强催化活性[17];值得注意的是,在碳基材料中异质原子的引入可有效地改变碳骨架周围电子分布状态,增加活性位点并有效调节碳材料的电化学行为,进而提高电化学反应效率[18-20]。
基于Fe3C优异的催化性能,同时结合异质原子掺杂碳材料具有高稳定性、良好导电性以及对于Fe3C纳米粒子包覆后可能的协同效应。本文拟采用二茂铁和PVP为原料,通过简单可控的水热法制备氧化铁纳米粒子均匀分散的聚合物凝胶[21],在尿素的辅助下,高温热解制备Fe3C/N-C纳米粒子,研究了热解温度对所制备纳米粒子微观结构的影响并探讨了其电催化析氢性能。本文将为开发廉价、高效且易于规模化生产的新结构碳基析氢材料探索新的技术途径。
1 实 验
1.1 样品的制备
1.1.1 试 剂
本实验中所用化学试剂如氢氧化钾、尿素、二茂铁、PVP、过氧化氢(H2O2)、无水乙醇等均为分析纯,来自于成都科隆化学品有限公司;聚四氟乙烯(PTFE)乳液来自于国药集团化学试剂有限公司;氩气(5N),来自于绵阳昌俊气体有限公司。
1.1.2 催化剂的制备
将5 g PVP和1 g二茂铁溶解在40 mL去离子水中,搅拌30 min后,加入1 mL H2O2(30 %质量分数),搅拌10 min,待溶液完全变成暗绿色后转移至50 mL反应釜中,在200 ℃下水热12 h后收集黑色布丁凝胶状产物,随后将凝胶在60 ℃下真空干燥48 h。将干燥后的产物研磨成粉末后,与尿素按照(质量比1∶1)充分混合,将一定量的混合物置于管式炉中,在氩气气氛保护下热解处理1 h。
1.1.3 工作电极的制备
将20 mg制备的催化剂置于烧杯中,加入一定量的PTFE乳液(10%质量分数)和无水乙醇(体积比1∶1),超声分散到呈现油墨状,然后将其取出并均匀涂抹至处理好的泡沫镍上(有效面积为1.0 cm2),最后置于60 ℃真空烘箱中干燥12 h即得到用于析氢性能检测的工作电极。
1.2 样品的性能及表征
采用德国X’Pert PRO型 X射线衍射仪分析材料的物相组成,德国Zeiss公司Sigma 500场发射扫描电镜观察材料的表面形貌,Libra透射电子显微镜分析材料的微观结构,英国雷尼绍公司Invia型激光拉曼光谱仪(激发波长514.5 nm)和美国赛默飞K-Alpha光电子能谱仪分析材料的组成成分。
析氢性能在上海辰华CHI760D工作站上进行表征,采用典型的三电极体系,其中负载产物的泡沫镍为工作电极、石墨为对电极、Hg/HgO为参比电极。以1.0 mol/L KOH为电解液,在测试线性扫描伏安曲线(LSV)之前,进行循环伏安(CV)活化测试,LSV测试之前进行IR补偿矫正,最后电势以可逆氢电势(RHE)表示:ERHE=EHg/HgO+0.098+0.059pH。
2 结果与讨论
2.1 凝胶产物的热重分析
图1为水热法所制备的凝胶材料及其与尿素混合物的热重曲线。从图中可以看出,纯凝胶材料的热分解行为可以分成5个过程:当温度低于145 ℃时(过程I),脱去少量的吸附水,导致约2%的失重;在145~265 ℃范围内(过程II)出现较大的失重(约33%),可能对应于凝胶材料内交联的部分PVP分子发生热分解;随着温度继续升高,在265 ~ 367 ℃范围内,样品出现约22%的质量损失(过程Ⅲ),这可能归因于为凝胶材料内PVP分子的进一步热分解;结合后面的XRD衍射结果,我们推断过程IV(367~458 ℃)应该为部分热分解后的PVP发生碳化的失重过程(约26%);最后,随着温度的增加,热失重逐渐变缓慢,到1200 ℃左右仅有约7%的重量损失,这可能对应着热解所得的石墨碳与所包裹的碳化铁纳米粒子发生碳热反应。尿素与凝胶混合物的热重曲线基本遵循相似的变化趋势,但仍存在两个变化:(1)过程II的起始温度显著增加至220 ℃左右,且该过程所对应的热分解更快速;(2)过程III和过程IV明显存在“合并”的行为,重量变化随温度变化更明显,这些变化可能是尿素参与凝胶材料内PVP分子热分解和碳化过程。鉴于此,为比较热解温度对所制备材料的结构、成分及电催化性能的影响,在高温热解退火过程中,我们采用了3个不同的温度(400、800和1 200 ℃)进行热解,对应样品分别命名为NC-400、NC-800和NC-1200。
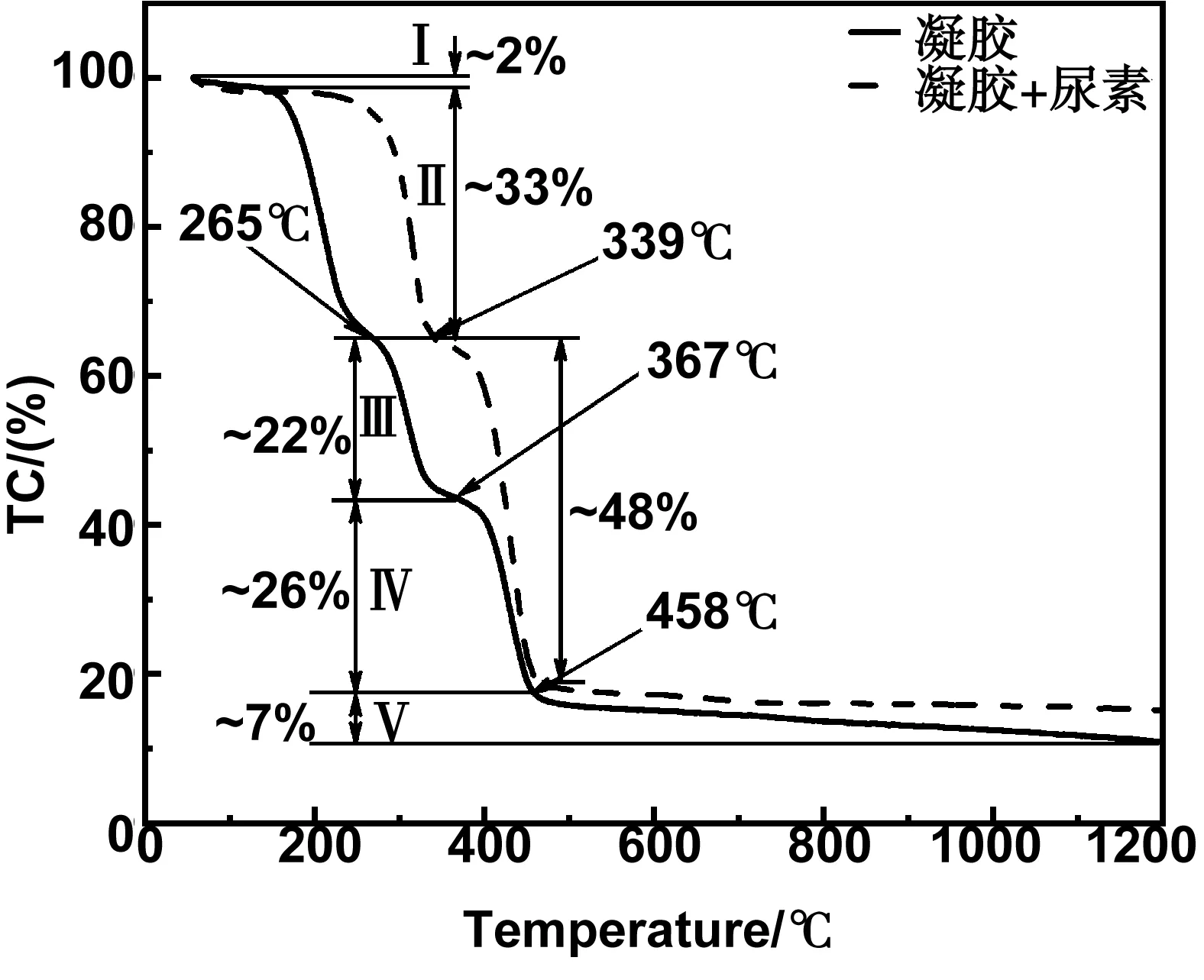
图1 凝胶、凝胶与尿素的混合物的热重曲线Fig 1 TGcurve of as-obtained gels and the mixture of gels and urea
2.2 催化剂的结构表征
图2(a)为不同热解温度下样品的X射线衍射花样(XRD)。在NC-400中,仅在26°左右出现一个较宽的衍射峰,表明产物中物相含有无定形碳,这是由于PVP分子在此温度下热解过程中交联碳化形成。当热解温度为800 ℃时,产物除了一定程度非晶化的石墨(002)峰外,在37.6°、39.8°、40.6°、42.8°、43.7°、45.0°、45.9 °、48.6°和49.1°出现了新的衍射峰,分别对应于Fe3C(JCPDS.35-0772)的(121)、(201)、(002)、(211)、(102)、(031)、(112)、(131)、(221)晶面,表明产物中存在石墨和Fe3C的复合相,这是由于在800 ℃下凝胶材料中存在的四氧化三铁(Fe3O4)纳米粒子与PVP热解产生的碳发生碳化反应所致。随着碳化温度增加至1 200 ℃后,产物除了石墨衍射峰和Fe3C衍射峰外,在44.6°和65°出现了尖锐的α-Fe(JCPDS.06-0696)的(110)和(200)晶面的衍射峰,而属于Fe3C的衍射峰明显减弱,表明在1 200 ℃下,Fe3C与临近的碳发生碳热反应被还原为铁(Fe)。值得注意的是,随着热解温度的增加,位于26°左右碳相的衍射峰变得越来越尖锐,表明从无定形碳到结晶态碳的转变。
众所周知,拉曼光谱被广泛应用于研究碳材料中杂化和结晶情况。如图2(b)所示,三个样品的拉曼光谱在1 355和1 585 cm-1处有明显的峰,分别对应于碳的D峰和G峰。D峰指一般为sp2杂化碳材料的无序或缺陷,G峰指结晶态石墨碳结构[22]。在NC-400中,D峰较宽,几乎不存在2D峰,表明NC-400中的碳为无定形碳,这与XRD结果一致;当热解温度为800 ℃时,D峰逐渐增强,表明了碳基质中存在氮掺杂而形成了结构缺陷;对于NC-1200,G峰增强并向左移动,表明样品中,sp2-C含量增加,证实了碳化反应诱导碳材料的石墨化程度加深,与XRD结果一致。
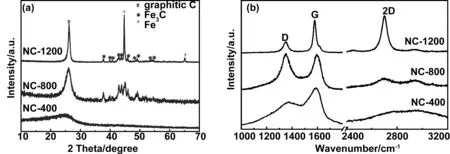
图2 不同热解温度下样品的XRD花样和拉曼光谱图Fig 2 XRD patterns and Raman spectra of as-obtained samples at different annealing temperature
采用扫描电镜和透射电镜观察了不同热解温度下制备样品的形貌。从图3(a)和(d)中可以看出,NC-400是一种由无定形碳堆积而成的块体结构,难以看出铁的氧化物纳米粒子出现,表明所生成的纳米颗粒均被PVP交联而成的无定形碳包裹在其中;NC-800呈现出球形纳米粒子聚集状,且尺寸分布较为均匀(如图3b);透射电镜给出了微观结构更多的细节信息,NC-800呈现出典型的实心核壳结构,粒径约为25~50 nm(如图3e)。对于NC-1200,从图3(c)和(f)中可以看出,进一步升高热解温度,发现其球形结构并没有被破坏,但纳米颗粒聚集堆叠,壳中碳层层数有所增加。上述结果充分表明,热解温度在Fe3C颗粒的形成中发挥着重要作用,不仅能控制复合材料中物相,形貌和结晶性,而且还能调控纳米粒子的分散性从而进一步调节其电催化性能。

图3 不同热解温度下所制备样品的(a-c)扫描电镜图(插图对应于放大图像)和(d-f)透射电镜图(a,d)NC-400,(b,e)NC-800,(c,f)NC-1200Fig 3 SEM and TEM images of as-obtained samples at different pyrolysis temperature. The insets correspond to enlarged images: (a,d) NC-400, (b,e) NC-800, (c,f) NC-1200
为了进一步表征样品的表面键合状态和化学组成,对NC-400,NC-800和NC-1200进行了X射线光电子能谱分析。从图4(a)中可以看出,NC-400、NC-800以及NC-1200的中含有C、N、Fe元素的存在,而由于氮的热不稳定性,在NC-1200中几乎没有N的含量。图4(b-A, b-B)中可以看出,NC-400和NC-800的C1s谱,对应于3种成键方式:位于284.8、285.3和286.0 eV的sp2-C、sp3-C以及C-N键[23-24],表明了氮掺杂碳的存在;而在NC-1200中仅含有sp2-C和sp3-C键,表明C-N键在高温下进行了分解。NC-400的N1s中,氮元素仅以吡啶氮的形式存在(如图4c-A)。图4b-B为NC-800的高分辨N1s谱,对应于氮的3种成键方式,401.0、400.0和398.5 eV的结合能反映了石墨氮、吡咯氮、吡啶氮的结合形式[23-24],进一步证明了氮元素掺入了碳材料的结构中。通过峰面积估算了吡啶氮、吡咯氮和石墨氮的含量,分别为38.74%、26.16%和35.10%。边缘缺陷位点吡啶氮能促进HER过程中H+的吸附和解吸过程,被认为是更有效的氮掺杂类型[25-26]。图4(d-A)展示出了NC-400的Fe2p图,位于709.9、710.7、724.4和726.4 eV的结合能分别对应于FeⅡ2p3/2、FeⅢ2p3/2、FeⅢ2p1/2和FeII2p1/2轨道能谱,匹配于Fe2+和Fe3+[27-28],证实了无定形碳中Fe3O4存在。Fe2p特征峰非常弱,与其他元素相比其含量几乎可以忽略不计,这表明Fe3O4几乎完全嵌入了碳基质中;图4(d-B)为NC-800的Fe2p图,相比于NC-400,Feε2p3/2和Feε2p1/2轨道卫星峰,表明NC-800中仅有Fe3+,证实了NC-800中含有Fe3C。图4(d-B)为NC-800的Fe2p图,位于710.7和724.4 eV的FeⅢ2p3/2和FeⅢ2p1/2轨道能谱,证实了NC-800中含有Fe3+,表明了Fe3C的存在。在NC-1200中,如图4d-C,Fe2p除了FeⅢ2p3/2和FeⅢ2p1/2轨道能谱之外,707.8和720.1 eV处两峰还揭示了Fe0的存在[28]。充分说明了随着温度升高在碳层内发生(Fe3O4→Fe3C→Fe-Fe3C)晶体转变,调整了产物的种类。
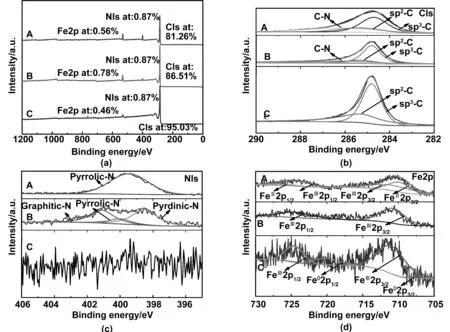
图4 (a)X射线光电子能谱,(b)高分辨C1s谱,(c)高分辨N1s谱,(d)高分辨Fe2p谱(A)NC-400, (B) NC-800, (C) NC-1200Fig 4 Survey XPS spectra,high-resolution C 1s XPS spectrum, high-resolution N 1s XPS spectrum, andFe 2p XPS spectra of samples: (A) NC-400,(B) NC-800, and (C) NC-1200
综上所述,我们可以推断出反应机理。当温度升高时,尿素和PVP快速分解交联形成无定形碳包覆的Fe3O4。随后升高温度,C-N键分解产生氮和碳的物种,Fe3O4纳米粒子与碳反应生成Fe3C,氮和碳物种在生成的Fe3C纳米颗粒的表面上形成了氮掺杂的碳层,从而形成Fe3C/N-C的核壳结构。最后,进一步升高温度,碳热反应的进行将碳层周围的Fe3C还原成Fe,最终形成Fe-Fe3C。
2.3 催化剂的电化学性能
我们研究了制备材料作为HER催化剂的催化性能,采用三电极体系测定了所制备材料在1.0 mol/LKOH溶液中的HER性能。图5(a)为不同样品的LSV极化曲线。对比三种样品可以看出,NC-400、NC-800、NC-1200在10 mA/cm-2电流密度下对应的过电位分别为302、211、272 mV。显然,NC-800具有最小的析氢过电位,表现出最好的HER催化性能,具有更高的催化活性。而NC-400具有最差的过电位(302 mV)与泡沫镍基底性能(305 mV)相差无几,这主要由于无定形块状样品中缺少暴露的活性位点造成的。为了更直观的评价Fe3C/N-C纳米复合结构的催化性能,将其与报道过的同类催化剂在表1中进行了比较。由表可知,所制备的 NC-800过电位较低,具备优异的电催化性能,可与文献所报道的同类催化剂性能媲美。
表1 Fe3C@N-C与先前报道的催化性能对比
Table 1 Comparison of the electrochemical catalytic performances of the recently reported catalysts with Fe3C@N-doped carbon

催化剂电解液过电位参考文献Fe3C/N-C1.0 mol/L KOH 211 mV[本项工作]Fe5C2-Fe3C@NC1.0 mol/L KOH209 mV[27]Fe/Fe3C@N-C0.5 mol/L H2SO4236 mV[28]Co@NCNT1.0 mol/L KOH244 mV[29]Co-N-C1.0 mol/L KOH224 mV[30]Fe3C/NC0.5 mol/L H2SO4288 mV[31]
众所周知,塔菲尔(Tafel)斜率是催化材料的固有性质,是权衡HER活性的另一重要参数,反映了HER反应速率以及催化反应的动力学过程[32]。图5(b)为对应的Tafel曲线,可以看出,NC-400、NC-800和NC-1200的Tafel斜率分别为199,137和178 mV/dec。显然,NC-800具有最小的Tafel斜率,在外加电位较低时,有较大的电流增益,有利于促进催化反应的进行[33],表明NC-800上的HER过程更加高效。此外,对NC-800在碱性电解质中连续10 h的稳定性测试。在图5(c)的i-t曲线中可以看出,在211 mV过电位下,电流响应随着时间的增加基本保持不变,证实了NC-800独特的结构可以在碱性电解液中保持较好的稳定性和耐久性。
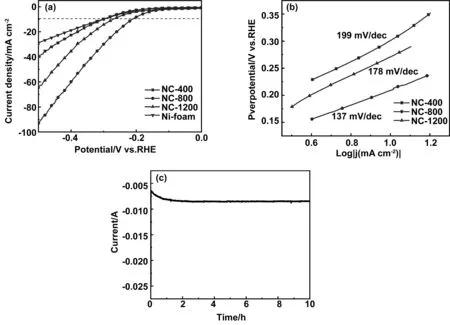
图5 (a)不同热解温度下样品的LSV曲线,(b)相应的Tafel曲线,(c)211 mV电位下NC-800的i-t曲线Fig 5 Linear sweep voltammograms of different carbonization temperature, the corresponding Tafel plots and i-t curves of NC-800 at 211 mV

图6 (a)NC-800的高分辨电镜,(b-e)NC-800的Mapping图,(a)中插图对应于电子衍射图Fig 6 Typical HRTEM image and mapping of NC-800.The inset in (a) is electron diffraction pattern image of NC-800
2.4 NC-800的高分辨电镜分析
为了进一步理解NC-800的催化机制,明确结构和元素组成,对其进行了高分辨电镜以及Mapping分析。图6a为NC-800的高分辨电镜图,从图中可以看出,单组分Fe3C纳米粒子呈球形分布,尺寸约为25~50 nm;此外,壳层由晶格间距约为0.34 nm的有序层组成,对应于碳的(002)晶面;核中约为0.21 nm的晶面间距匹配于Fe3C的(211)晶面,氮掺杂碳以纳米薄层形式紧密包覆于纳米Fe3C粒子表面。在连续进行的催化反应中,核壳结构能有效地避免单组分Fe3C出现团聚现象。同时,多层氮掺杂碳包裹的Fe3C纳米粒子有效抑制了催化剂的失活,有利于提升NC-800的催化活性和稳定性。选区电子衍射图(图6a插图)充分说明了NC-800是一种多晶结构。如图6b-6e,Mapping图像进一步证实了Fe集中分布于球形粒子中间,C、N均匀分布于整个区域。以上结果表明,采用水热结合800 ℃热解成功制备了氮掺杂碳包裹的碳化铁纳米粒子。
综上,我们认为HER催化活性提高的主要原因主要为:首先,高电负性杂原子的掺杂形成的吡啶氮和石墨氮提供了强有力的活性位点,提高催化活性;其次单分散的Fe3C纳米粒子与氮掺杂碳层之间的协同效应共同促进催化活性的提升;最后典型的核壳结构不仅显著地提升了Fe3C/N-C的导电性,创造了活性位点,提高了电催化活性,还保证了Fe3C核的化学和结构稳定性。
3 结 论
本文采用简单的水热-热解两步法,成功制备了Fe3C/N-C的复合纳米粒子,发现热解温度在Fe3C/N-C的形成中发挥着重要作用。此外,表征了所制备催化剂在碱性溶液中的催化性能。结果表明,Fe3C/N-C的复合纳米粒子表现出优异的HER性能(211 mV@10 mA/cm2)以及较小的Tafel斜率(137 mV/dec),同时在碱性电解液中表现出优越的稳定性。Fe3C/N-C纳米粒子优异的催化性能归因于过渡金属碳化物和氮掺杂碳的协同效应以及其独特的核壳结构。该工作将为过渡金属碳化物和氮掺杂碳的复合结构调控提供新思路,为发展高效廉价的非贵金属电催化剂提供技术支撑。
猜你喜欢
化工管理(2022年14期)2022-12-02
昆明医科大学学报(2022年1期)2022-02-28
陶瓷学报(2021年1期)2021-04-13
军事文摘(2020年20期)2020-11-16
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
中国特种设备安全(2020年11期)2020-06-09
上海建材(2020年12期)2020-04-13
中学生数理化·八年级物理人教版(2020年12期)2020-01-01
中学生数理化·八年级物理人教版(2018年12期)2019-01-31
