
2-甲基-3-硝基苯乙腈合成工艺改进
2020-06-30 05:59李匡元李娟张怡门靖
化工与医药工程 2020年2期
李匡元,李娟,张怡,门靖
(1.陕西万盛健康产业有限公司,陕西西安 710000;2.西安万隆制药股份有限公司,陕西西安 710119)
罗匹尼罗(Ropinirole),化学名称:4-(2-二正丙基胺乙基)-1,3-二氢-2H-吲哚-2-酮,结构式见Chart 1,CAS号:91374-21-9,分子式:C16H24N2O,分子量:260.375 g/mol,商品名:Requip。该药物由英国葛兰素史克(GlaxoSmithKline,GSK)研发,于1997年首次被批准用于帕金森病(Parkinson's Disease,PD),后被批准用于治疗中度到重度的不宁腿(多动腿)综合症(Restless Legs Syndrome,RLS)[1-4]。罗匹尼罗的药理作用是作为多巴胺受体激动剂(Dopamine Agonist),增强多巴胺的作用,矫正中枢神经传导素的不平衡,解除症状并维持病人的自主性与活动力[5-6]。
2-甲基-3-硝基苯乙腈是合成罗匹尼罗原料药的关键中间体,已报道的合成路线依据起始物料主要有两种路线。路线一是以邻二甲苯为起始原料,经硝化、氧化、水解、还原、氯化和氰化等6 步反应制备[7]。路线合成工艺如图1所示,该反应步骤冗长,同时涉及硼氢化钠、有毒品三氯化磷的使用;该工艺引入氰基的原理是通过氯代烃与剧毒品氰化钾反应获得的,使用大量的氰化物必然会污染环境和造成人身伤害,危险性大,废液处理困难,总收率仅3.3%,成本较大,不适合工业放大生产。
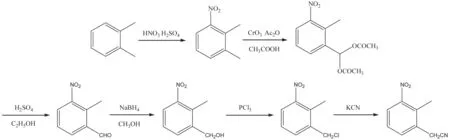
图1 2-甲基-3-硝基苯乙腈合成路线一Fig.1 The first synthesis route of 2-methyl-3-nitrophenylacetonitrile
路线二采用上述路线中间体2-甲基-3-硝基苯甲醛为起始原料,通过还原、氯代和氰代三步反应制备[8]。路线合成工艺如图2所示,该合成路线的优势是针对繁琐的实验合成步骤进行了优化,缩短了实验工期和设备成本,同时采用二氯亚砜替代三氯氧磷,避免大量废液的产生。但是仍存在诸多不足,还原步骤反应液需要分馏,再者不可避免地使用剧毒品氰化物,限制其工业化生产。
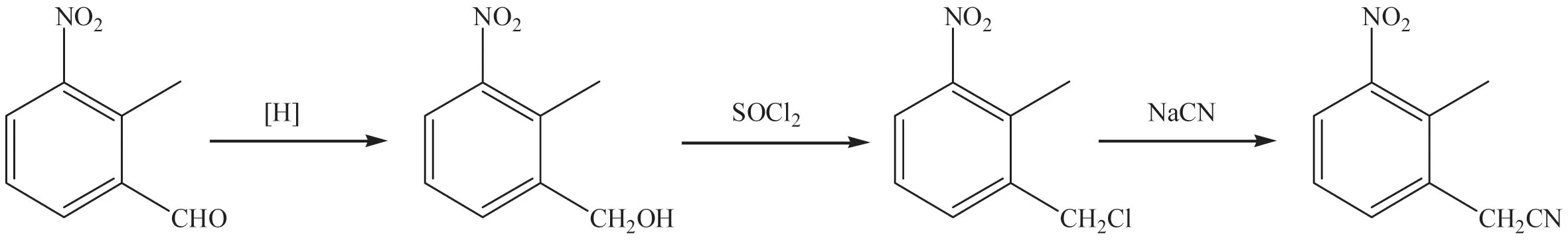
图2 2-甲基-3-硝基苯乙腈合成路线二Fig.2 The second synthesis route of 2-methyl-3-nitrophenylacetonitrile
纵观现有合成路线,目标产品的合成关键是氰基的引入,针对现有工艺不足,本文提出了以酰胺脱水来获得氰基官能团的思路,避免了传统氰化物的使用[9-10]。提出了改进的合成路线:以市场供应充足的2-甲基苯乙酸为原料,经硝化、酰胺化、脱水三步反应制备目标产品2-甲基-3-硝基苯乙腈,其结构经1H NMR和MS 确证。实验中以正交试验为研究方法,对工艺进行了优化改进,以期获得一条操作简单、工艺稳定、设备匹配性好的合成路线,对2-甲基-3-硝基苯乙腈的工业化生产及下游原料药的研究提供借鉴意义。
1 实验部分
1.1 仪器和试剂
仪器:AV-400 MHz型核磁共振仪(氘代二甲基亚砜DMSO-d6为溶剂,德国Bruker公司),3100型质谱仪(电喷雾质谱法ESI-MS,美国Waters公司),LC-2010型液相色谱仪(高效液相色谱法HPLC 测定,日本岛津)。
试剂:2-甲基苯乙酸(含量>98.0%),上海启仁化工有限公司;三氟乙酸酐(含量>99.0%),衢州市瑞尔丰化工有限公司;浓硫酸、硝酸、二氯甲烷、三乙胺、二氯亚砜、氨气、无水硫酸镁(分析纯,国药集团化学试剂有限公司)。氮气(西安欣祥电气有限责任公司,工业级)。
1.2 合成原理与工艺路线
合成原理:以2-甲基苯乙酸为原料,经硝化、酰胺化、脱水三步反应制备目标产品2-甲基-3-硝基苯乙腈,合成工艺路线如图3所示。对每一步合成工艺进行了优化,实验重点讨论了二氯亚砜和中间体(3)的摩尔比、氨气和中间体(3)的摩尔比、反应时间三种因素对制备中间体(4)的影响、以及脱水试剂、三氟乙酸酐和化合物(4)的摩尔比、反应温度、pH值对目标产品(1)反应收率的影响。
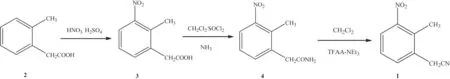
图3 2-甲基-3-硝基苯乙腈合成工艺改进Fig.3 Improvement of synthetic process of 2-methyl-3-nitrophenylacetonitrile
1.3 实验步骤
1.3.1 中间体(3)的合成工艺描述
向3L反应瓶中通氮气30 min,加入起始原料2- 甲基苯乙酸610 g(4.06 mol),分批加入混酸722.0 mL(284.0 mL 硝酸和438.0 mL 浓硫酸),搅拌30 min,反应液经冰盐浴降温至0~10℃,搅拌1 h,静置,分层,有机相经120 mL 饱和碳酸氢钠水溶液洗涤,搅拌析晶2 h,过滤得到类白色固体,即中间体(3)635.0 g,收率79.8%。1H NMR(400 MHz,CDCl3为溶剂)δ:2.36(s,3H,Ph-CH3),3.50(s,2H,-CH2COOH),10.92(s,1H,-COOH),7.92(d,J=4.8 Hz,1H),7.26(d,J=4.0 Hz,1H),7.32(d,J=4.0 Hz,1H);MS(ESI)m/z: 196.2{ [M+H]+}。
1.3.2 中间体(4)的合成工艺描述
向3L反应瓶中通氮气30 min,加入1.8L二氯甲烷和中间体(3)600.0 g(3.06 mol)搅拌30 min,开始滴加二氯亚砜,滴加过程中保持反应体系温度不高于25℃,滴加完毕后升温至回流反应2.0 h,反应液冷却、浓缩掉约一半体积反应液,再通入氨气,反应体系由酸性转变为碱性,常温反应2.0 h,过滤,加入二氯甲烷1.0 L 进行萃取,萃取3次,每次萃取搅拌0.5 h,静止分层0.5 h,有机相合并,经无水硫酸钠0.8 kg 搅拌干燥3 h,过滤、滤液减压浓缩(热水温度30~35℃、真空度≤-0.08 MPa)至冷凝管不滴出液体,得粗品543.2 g。经打浆纯化得519.2 g中 间 体(4),收 率86.5%。1H NMR(400 MHz,CDCl3为溶剂)δ:2.36(s,3H,Ph-CH3),3.54(s,2H,-CH2CONH2),6.04(s,2H,-NH2),7.90(d,J=4.8 Hz,1H),7.24(d,J=4.0 Hz,1H),7.30(d,J=4.0 Hz,1H);MS(ESI)m/z: 195.2{ [M+H]+}。
1.3.3 目标产品(1)的合成工艺描述
向3L反应瓶中通氮气30 min,依次加入四氢呋喃1.8L和中间体(4)510.0 g(2.62 mol),搅拌,待降温至-10~-5℃后,加入622.0 g 三乙胺、770.8 g(3.67 mol)三氟乙酸酐,保温40℃反应,反应结束后,向体系中加入2 M 碳酸氢钠水溶液调节体系pH值至10,再加入乙酸乙酯1.2 L 萃取(每次0.4 L,提取三次),有机相经无水硫酸钠干燥3 h,过滤、浓缩得到粗品,再经重结晶纯化得到目标产品1 383.0 g,为白色粉末状固体,收率82.6%,反应总收率57.0%。产品纯度为99.6%(HPLC 法,含C9H8N2O2不得低于99.0%)。干燥失重为0.4%(按照中国药典2015年版四部通则0831 干燥失重测定法进行试验,不得大于0.8%)。水分为0.3%(参照中国药典2015年版四部通则0832 水分测定法第一法,不得大于0.6%)。
2 结果与讨论
2.1 中间体(4)工艺优化
实验通过L9(34)正交实验法,以反应收率为评价指标,设计探讨了二氯亚砜和中间体(3)的摩尔比(A)、氨气和中间体(3)的摩尔比(B)、反应时间(C)三个影响反应收率的主要因素,每个因素各取3个水平,优选出最佳工艺参数。正交实验设计如表1所示,实验结果如表2所示。
由上述直观分析和极差分析结果可知,各实验因素对中间体(3)反应收率的影响程度依次为B>A >C,即氨气和中间体(3)的摩尔比影响显著,二氯亚砜和中间体(3)的摩尔比和反应时间影响次之。结合各因素位极之和K值可以看出,合成化合物(4)的最佳工艺组合为A2B3C1,即最佳反应条件为:n(二氯亚砜)∶n(氨气)∶n(3)=1.3∶1.4∶1,反应时间2 h时收率最高。在此条件下,重复3次实验,平均收率为86.3%。
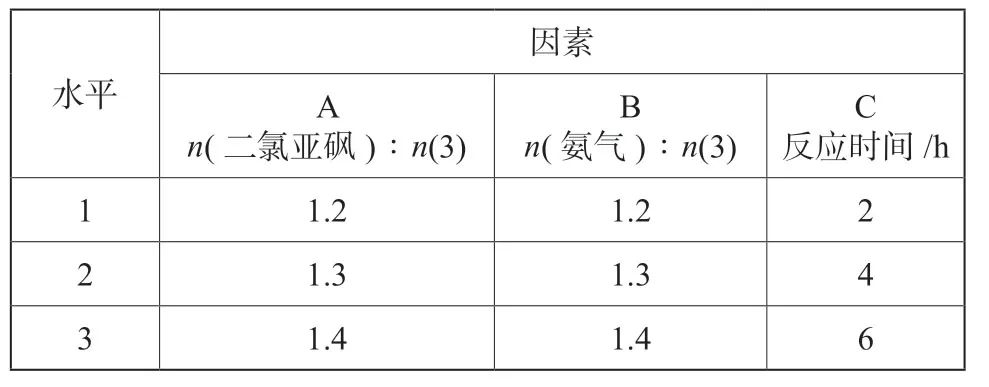
表1 化合物(4)正交设计实验Tab.1 Orthogonal experiment design of compound(4)
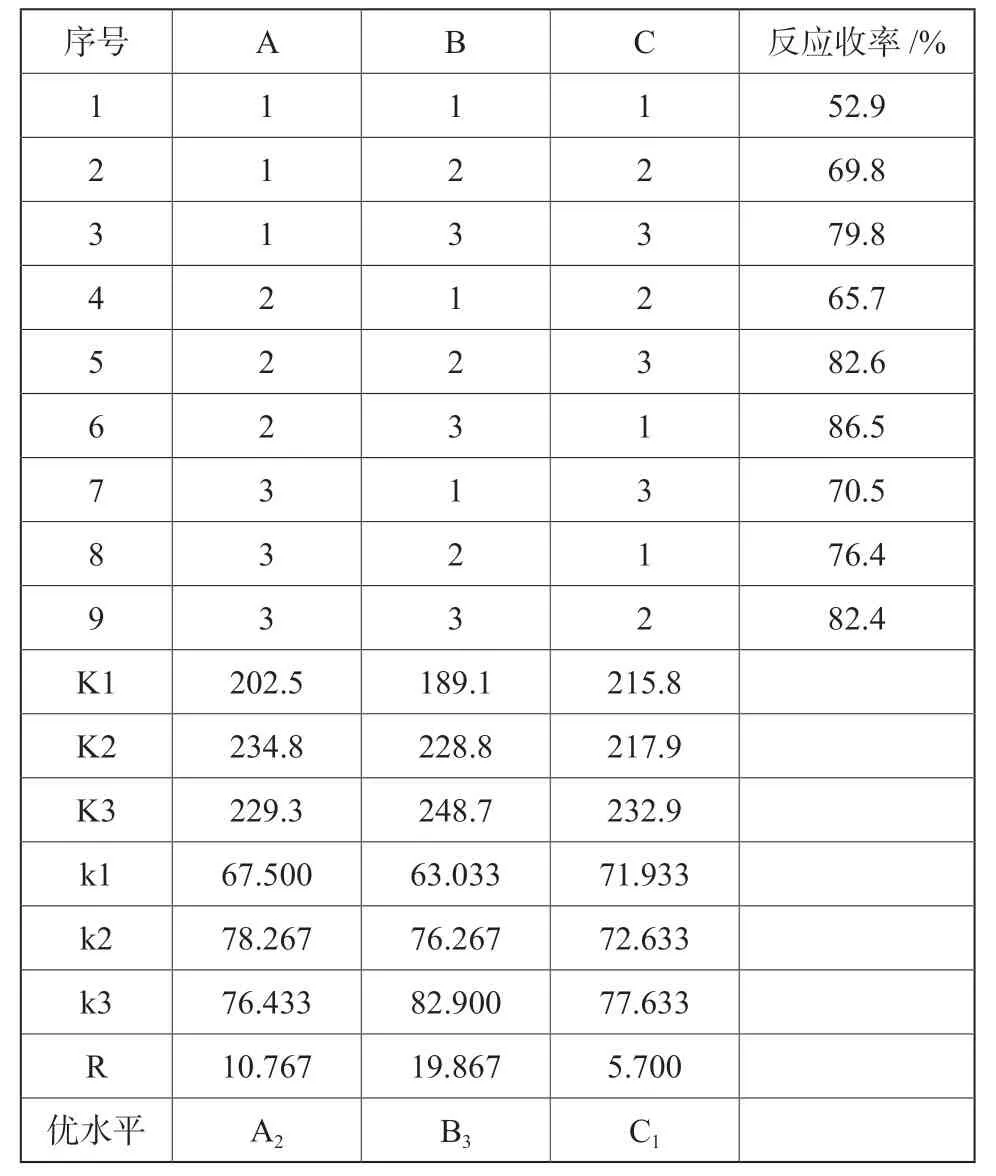
表2 化合物(4)正交实验结果Tab.2 Results of orthogonal test of compound(4)
实验中发现,向反应体系通入氨气时,氨气用量少则反应慢,化合物(2)约有质量分数为10%的剩余,反应不充分;氨气用量较多,虽反应时间有所缩短,但后处理会产生大量碱性废液,给环保带来巨大压力,优化实验表明当氨气用量n(氨气)∶n(3)= 1.4∶1时反应收率较高,与正交试验结果相一致。
2.2 目标产品(1)工艺优化
2.2.1 脱水剂的筛选与优化
芳香类酰胺在P2O5、POCl3、SOCl2等脱水剂存在下制备相应的腈是获得腈化合物的经典方法,前期我们尝试了这些酸性脱水剂的效果,发现反应较慢、杂质多,而且后处理会产生大量酸性废液,不利于环保。继而考察了三氟乙酸酐-三乙胺(TFAA-NEt3)、甲烷磺酰氯(CH3SO2Cl)两种温和脱水剂的反应效果[11-12]。实验发现采用甲烷磺酰氯作脱水剂时,需要用到盐酸洗涤过滤,对设备要求严格,中间体(4)约有5%~8%的残留,反应不完全。故我们选择三氟乙酸酐-三乙胺作为脱水剂,实验中选取反应温度区间30~60℃进行优化实验,测试结果表明反应温度40℃时反应收率比较稳定,为81%~84%,效果较优。该脱水剂使用过程中工艺操作简单、反应条件温和。
2.2.2 正交试验结果
实验通过L9(34)正交实验法,以反应收率为评价指标,设计探讨了三氟乙酸酐和化合物(4)的摩尔比(A)、反应温度(B)、pH值(C)三种因素对反应收率的影响,优选出最佳工艺参数。正交实验设计如表3所示,实验结果如表4所示。
由上述直观分析和极差分析结果可知,各因素对反应收率的影响程度依次为A >B >C,即三氟乙酸酐和化合物(4)的摩尔比对反应影响更为显著,反应温度和pH值影响次之,合成目标产品(1)的最佳工艺组合为A2B2C3,即最佳反应条件为∶n(三氟乙酸酐)∶n(4)=1.4∶1,反应温度40℃及pH值为10时,反应收率最高,目标产品HPLC 纯度达99.6%。在此条件下,重复3次实验,平均收率为82.2%。
2.3 中试极限试验
对目标产品的干燥条件进行摸索,并以大生产为指导原则,测试了目标产品的湿品于40~50℃干燥1~14 h的情况,考察干燥条件对主峰含量、干燥失重和水分等指标的影响,测试结果如表5所示。

表3 目标产品(1)正交设计实验Tab.3 Orthogonal experiment design of target product(1)
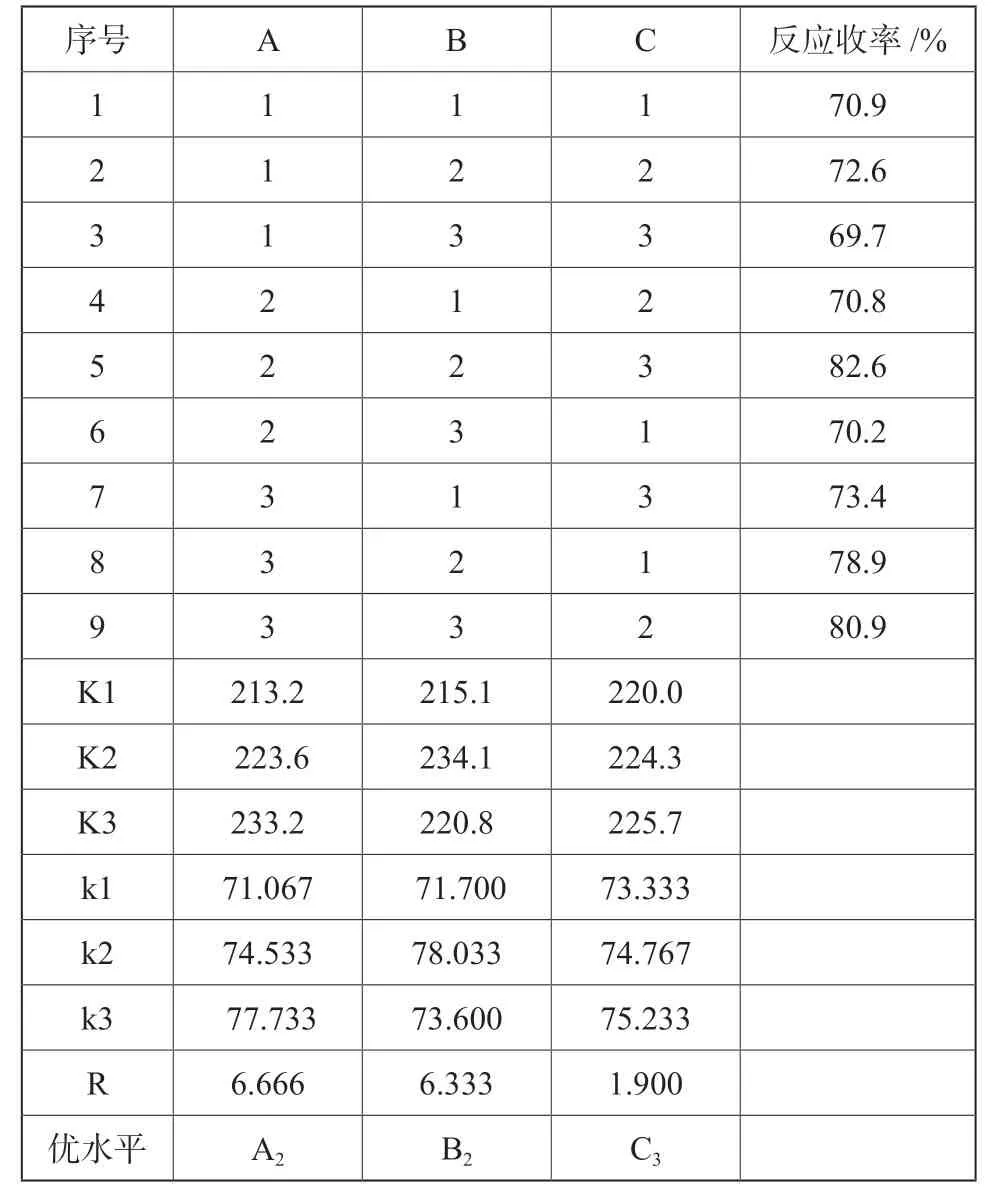
表4 目标产品(1)正交实验结果Tab.4 Results of orthogonal test of target product(1)
由表5可知,干燥时间短,目标产品的主峰含量较低、干燥失重和水分均较大。延长干燥时间,测试指标均明显提升。干燥10 h时目标产品的主峰含量最高为99.6%,且干燥失重为0.4%,水分为0.3%,均符合质量标准。干燥时间12 h时虽主峰含量略有降低,但符合质量标准,若继续延长干燥时间,产品主峰含量降低至98.2%,不符合质量标准。故中试极限条件为湿品45~55℃,干燥10~12 h。
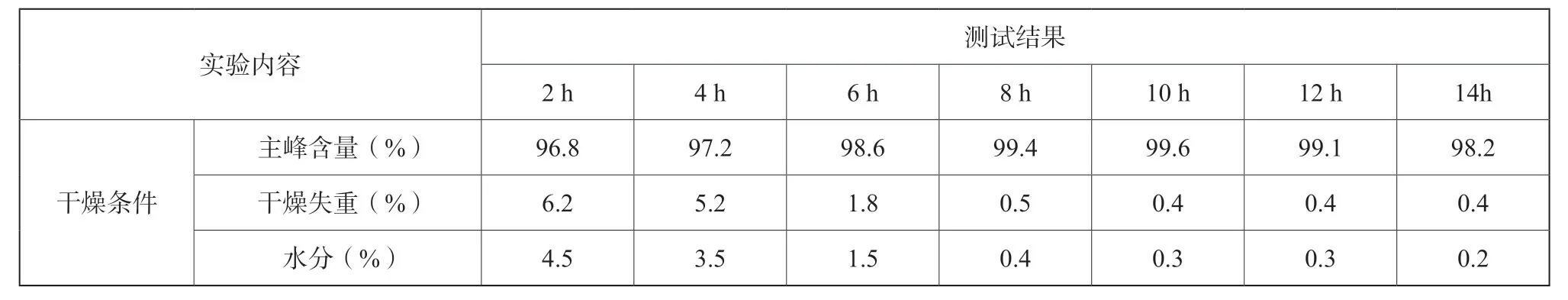
表5 干燥条件对目标产品的影响Tab.5 Effect of drying condition on target product
3 结论
以廉价易得的2-甲基苯乙酸为原料,经硝化、酰胺化、脱水三步反应成功制备目标产品2-甲基-3-硝基苯乙腈。采用正交试验分析合成中的关键参数,获得了适宜的生产工艺∶n(二氯亚砜)∶n(氨气)∶n(3)=1.3∶1.4∶1,反应时间2 h;三氟乙酸酐-三乙胺作脱水剂,且n(三氟乙酸酐)∶n(4)=1.4∶1,反应温度40℃及pH值为10。该工艺路线反应总收率57.0%,纯度99.6%,满足医药中间体高品质的要求。并对目标产品的干燥中试极限条件进行了探讨,湿品40~50℃,干燥10~12 h,为2-甲基-3-硝基苯乙腈的工业化生产及下游原料药的研究提供借鉴意义。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
云南化工(2022年9期)2022-10-12
中国药学药品知识仓库(2022年10期)2022-05-29
能源化工(2021年6期)2021-02-26
汕头大学学报(自然科学版)(2020年4期)2020-12-14
渔业现代化(2019年3期)2019-07-11
现代农业科技(2018年23期)2018-02-18
股市动态分析(2015年12期)2015-09-10
安徽农业科学(2015年9期)2015-01-12
