
基于CRISPR/Cas9技术构建NPHP1基因敲除的肾集合管上皮细胞
2020-06-29 05:10陈华木孙良忠胡妙月林宏容王海燕钟静琳吴小红
山东医药 2020年17期
陈华木,孙良忠,胡妙月,林宏容,王海燕,钟静琳, 吴小红
1 中山大学附属第一医院,广州510080;2 中山大学孙逸仙纪念医院;3 南方医科大学南方医院
肾单位肾痨(NPHP)是一种慢性间质性囊肿性肾病,是导致儿童终末期肾脏病(ESRD)最常见的遗传性疾病[1,2]。NPHP属于常染色体隐性单基因遗传病,目前发现的致病基因有20种(NPHP1~20)[3,4]。NPHP1是最常见的致病基因,编码蛋白nephrocystin-1,主要表达在肾脏远曲小管和集合管上皮细胞中[5]。在明确致病基因突变的NPHP患者中,NPHP1突变占50%~60%[5~7];NPHP1突变的主要类型是大片段缺失,少数为点突变或杂合缺失伴点突变[8,9]。目前,NPHP的发病机制尚未十分清楚。CRISPR/Cas9技术是近年开发的简单高效基因编辑技术,其原理是通过单一引导RNA序列(sgRNA)识别特定靶DNA序列,将内切酶Cas9引导至靶DNA的设定位置进行切割,使DNA双链断裂,细胞在通过自身非同源性末端修复(NHEJ)时,可使目标区域DNA出现插入、缺失等改变,达到基因敲除或改造的目的[10]。2016年1月~2018年3月,我们利用CRISPR/Cas9基因编辑技术在体外构建NPHP1基因敲除的犬肾集合管上皮细胞,为深入研究NPHP的发病机制和NPHP1基因的功能提供细胞模型。
1 材料与方法
1.1 主要实验材料 犬肾集合管上皮细胞MDCK(余学清教授惠赠);PX459质粒(Addgene,62988,美国);Stbl3感受态细胞(全式金,CD521,北京);无内毒素质粒小提中量试剂盒(天根生化科技有限公司,DP118,北京);DNA提取试剂盒(天根生化科技有限公司,DP318,北京);质粒PX459测序引物U6-Fwd:5′-GAGGGCCTATTTCCCATGATTCC-3′;兔抗NPHP1多克隆抗体(Sigma,SAB2104055,美国);鼠抗β-Actin单克隆抗体(CST,3700,美国);羊抗兔HRP酶标二抗(CST,7074,美国);马抗鼠HRP酶标二抗(CST,7076,美国);反转录第一链cDNA合成试剂盒(Thermo Fisher,K1622,美国);实时定量PCR扩增仪(TB GreenTMPremix Ex TaqTMⅡ,Takara,RR820A,日本);本研究的引物合成、sgRNA合成和基因测序均由广州华大基因有限公司完成。
1.2 sgRNA的设计及合成 利用CRISPR在线设计工具(http://crispr.mit.edu),在犬NPHP1基因(Gene ID:403780)的第5外显子(SH3功能结构域的起始部分),设计3对sgRNA(表1),分别为sgRNA5、sgRNA6、sgRNA7;在sgRNA正义链(F)的5′端添加CACC、反义链(R)的5′端添加AAAC形成粘性末端,可与BbsⅠ酶酶切后的粘性末端互补。

表1 CRISPR/Cas9 敲除MDCK细胞NPHP1的sgRNA序列
1.3 重组真核表达载体PX459-sgRNA的构建 使用限制性内切酶BbsⅠ酶消化1 μg质粒PX459,使质粒PX459形成线性载体。将sgRNA5~7的F、R在T4多聚核苷酸激酶作用下磷酸化和退火形成sgRNA二聚体。将sgRNA二聚体与PX459线性载体在T4 DNA连接酶作用下连接,构建重组真核表达载体PX459-sgRNA。用质粒保护的核酸外切酶去除非特异性连接,并转化到Stbl3感受态细胞中;将感受态细胞均匀涂到含氨苄青霉素的LB/Amp琼脂平板培养基上,过夜培养后挑选单菌落;摇菌培养后进行菌液PCR反应(上下游引物分别为U6-Fwd和sgRNA-R),结果阳性者进行质粒抽提并进行测序验证及双酶切鉴定。
1.4 MDCK细胞的培养及质粒转染 使用DMEM/F12培养基常规培养,在转染前1 d将生长状态良好的MDCK细胞以2×104/孔接种于24孔板中;当细胞融合度达70%~90%时,将细胞分为三组进行质粒转染实验;阴性对照组不转染;空载组和转染组每孔分别转染空质粒或重组质粒PX459-sgRNA 500 ng+Lipo2000 0.5 μL。转染8 h后补充无血清DMEM/F12培养基500 μL/孔,放回37 ℃、5% CO2培养箱中继续培养。
1.5 敲除NPHP1基因MDCK细胞的单克隆筛选 在重组质粒转染MDCK细胞48 h后,开始加入嘌呤霉素3 μg/mL;每天换液1次,换液时加入嘌呤霉素3 μg/mL;持续5 d或观察至阴性对照组和空载组均无细胞生长时停止嘌呤霉素筛选,改用DMEM/F12完全培养基(含10%胎牛血清、100 U/mL青霉素、0.1 mg/mL链霉素)培养。待嘌呤霉素筛选后的细胞恢复正常生长时,通过有限稀释法将细胞接种到96孔板中继续培养;7~14 d后观察96孔板,对存在细胞的孔做标记;将里面的细胞转移到24孔板中,扩大培养后依次转移到6孔板和细胞培养瓶中。
1.6 敲除NPHP1基因MDCK细胞的基因测序鉴定 使用DNA提取试剂盒对筛选到的细胞分别提取DNA,运用Primer Premier 5软件对犬NPHP1的第5外显子区域设计PCR测序引物。NPHP1-5F:5′-AATA-CAGGTCCCATAGTCTCA-3′;NPHP1-5R:5′-ATGCAAAC-TCATCCTTAGGTC-3′。退火温度为55 ℃,产物大小为582 bp。PCR扩增后,将产物送检测序。
1.7 敲除NPHP1基因MDCK细胞的脱靶检测 通过CRISPR在线设计工具(http://crispr.mit.edu)设计sgRNA时,根据网站生成的可能脱靶位点(8个),在犬基因组中找到对应位置,根据上下游序列设计引物(表2),通过PCR扩增和Sanger测序分析脱靶情况。

表2 CRISPR/Cas9敲除MDCK细胞NPHP1脱靶位点检测的上下游引物、退火温度及产物大小
1.8 通过shRNA敲低NPHP1基因MDCK细胞的构建 针对犬NPHP1的mRNA序列(NM_001195841.1),设计shRNA干扰位点(5′-CTGCACCCATCGGGAACTATA-3′)与慢病毒载体GV493一起构建重组慢病毒载体,由上海吉凯基因化学技术有限公司完成。通过慢病毒转染shRNA进MDCK细胞后,经5 μg/mL嘌呤霉素筛选14 d后,成功构建NPHP1敲低MDCK细胞。
1.9 MDCK细胞中NPHP1 mRNA检测 采用RT-qPCR法。收集MDCK细胞及CRISPR/Cas9技术敲除NPHP1基因的MDCK细胞,通过TRIzol法提取细胞总RNA,反转录合成cDNA,以cDNA为模板进行实时定量PCR扩增。引物序列:NPHP1-F:5′-AGCAGGAGGGGAAGAAGT-3′,NPHP1-R:5′-TGGAAGAGTGTGGAAGGC-3′,产物大小为144 bp;GAPDH-F:5′-AAGGTAGTGAAGCAGGCA-3′,GAPDH-R:5′-GGTGGAAGAGTGGGTGTC-3′,产物大小为105 bp。反应条件:95 ℃预变性30 s;95 ℃变性5 s,60 ℃退火30 s,扩增40个循环。以GAPDH为内参,根据2-ΔΔCt计算NPHP1的mRNA相对表达量。ΔΔCt=(CT实验组目的基因-CT实验组内参)-(CT对照组目的基因-CT对照组内参)。
1.10 MDCK细胞中nephrocystin-1蛋白检测 采用Western blotting法。收集MDCK细胞及CRISPR/Cas9、shRNA技术敲除(敲低)NPHP1基因的MDCK细胞,PBS缓冲液洗涤3次;RIPA细胞裂解液(含PMSF蛋白酶抑制剂)冰上裂解10 min,超声破碎仪使蛋白与核酸彻底分离;4 ℃下以12 000 r/min离心15 min去除沉淀,加入5×Loading Buffer在100 ℃下变性15 min;电泳,转膜,5%脱脂奶粉封闭2 h;加一抗(NPHP1 1∶1 000,β-Actin 1∶1 000)4 ℃孵育过夜,PBST缓冲液洗膜;二抗(1∶5 000)室温孵育1 h,PBST缓冲液洗膜;ECL化学发光液曝光显影,观察蛋白表达情况。
1.11 MDCK细胞形态观察 收集MDCK细胞及CRISPR/Cas9、shRNA技术敲除(敲低)NPHP1基因的MDCK细胞,换液时用PBS冲洗2~3次,添加完全培养基,倒置显微镜下观察细胞形态,同时拍照记录。

2 结果
2.1 重组真核表达载体PX459-sgRNA构建情况 对含重组真核表达载体的菌液行PCR扩增,均可见清晰条带(目标产物大小为273 bp)。质粒PX459单酶切后,可见1个线性质粒条带(大小约10 000 bp),双酶切后可见2 个条带;而重组真核表达载体单酶切均未见线性质粒条带,双酶切可见1 个条带,为线性质粒条带。基因测序结果发现,重组真核表达载体相对质粒PX459均插入了一段序列,均与对应sgRNA-F序列一致。以上结果表明,重组真核表达载体构建成功,分别为PX459-sgRNA5、PX459-sgRNA6、PX459-sgRNA7(图1,见彩插Ⅰ)。

注:A为重组真核表达载体PX459-sgRNA菌液PCR凝胶电泳图,其中①、②、③表示PX459-sgRNA5,④、⑤、⑥表示PX459-sgRNA6,⑦、⑧、⑨表示PX459-sgRNA7,重组真核表达载体菌液PCR的产物大小为273 bp;B为重组真核表达载体PX459-sgRNA酶切鉴定图,其中1、2、3、4分别表示质粒PX459和重组真核表达载体PX459-sgRNA5、6、7,每组各3个孔,每组a、b、c分别表示未酶切、单酶切和双酶切;C~F为质粒PX459与重组真核表达载体PX459-sgRNA的Sanger测序图,其中C表示质粒PX459,D、E和F分别表示重组载体PX459-sgRNA5、6、7,黑线标记分别为sgRNA5、6、7所在位置。
图1 重组真核表达载体PX459-sgRNA扩增、酶切鉴定和基因测序验证结果
2.2 NPHP1基因敲除MDCK细胞的敲除位点Sanger测序结果 Sanger测序结果显示,同源DNA双链均发生了非3N移码突变。第一链从sgRNA5的第18位点(chr17:-35122766)到sgRNA7的第17位点(chr17:-35122715)之间缺失52个碱基,第二链在sgRNA5的第17位点(chr17:-35122767)缺失1个碱基(图2,见彩插Ⅰ),分别为c.500_551del(p.His167GlnfsTer14)和c.499delC(p.His167ThrfsTer31)。
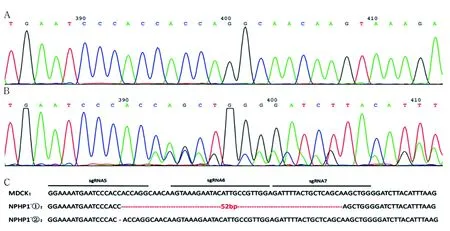
注:A:MDCK细胞;B:NPHP1基因敲除MDCK细胞;C:MDCK细胞与NPHP1基因敲除MDCK细胞基因序列比对图;NPHP1-①和NPHP1-②为NPHP1基因敲除株的双链DNA序列;sgRNA5~7为靶向NPHP1基因第5外显子的3条sgRNA碱基序列。
图2 NPHP1基因敲除MDCK细胞的敲除位点Sanger测序结果
2.3 NPHP1基因敲除MDCK细胞的脱靶情况 Sanger测序显示,NPHP1基因敲除MDCK细胞和MDCK细胞对比,这8个可能脱靶位点的基因序列均一致,未出现插入缺失等基因突变情况,提示上述可能脱靶位点均未出现脱靶情况。
2.4 MDCK细胞NPHP1 mRNA表达情况 RT-qPCR结果显示,NPHP1基因敲除MDCK细胞NPHP1 mRNA表达量为0.62±0.14,低于MDCK细胞的1.01±0.20(P<0.01)。
2.5 MDCK细胞nephrocystin-1蛋白表达情况 CRISPR/Cas9技术构建的NPHP1基因敲除MDCK细胞nephrocystin-1表达完全消失,通过慢病毒转染shRNA敲低NPHP1基因MDCK细胞的nephrocystin-1表达减低。见图3。

注:A、B为MDCK细胞,C、D为CRISPR/Cas9技术敲除NPHP1基因的MDCK细胞,E、F为shRNA敲低NPHP1基因的MDCK细胞,β-actin为内参。
图3MDCK细胞中nephrocystin-1蛋白表达情况(Western blotting法)
2.6 不同MDCK细胞的细胞形态 MDCK细胞聚集分布,呈铺路石样改变。CRISPR/Cas9敲除NPHP1基因的MDCK细胞外形有所改变,与通过慢病毒转染shRNA敲低NPHP1表达的MDCK细胞外形改变类似,略呈长梭形,细胞间隙稍增大(图4,见彩插Ⅰ。)

注:A为MDCK细胞,B为shRNA敲低NPHP1基因的MDCK细胞,C为CRISPR/Cas9技术敲除NPHP1基因的MDCK细胞。
图4 不同MDCK细胞的细胞形态(倒置显微镜,400×)
3 讨论
NPHP是儿童青少年肾衰竭的主要遗传性疾病,发病机制目前尚未十分清楚。NPHP1是最主要的致病基因。既往研究主要是通过RNAi技术敲低NPHP1表达来研究NPHP1基因的功能。我们前期对NPHP1表达降低诱导MDCK细胞发生上皮细胞间-充质转分化及与ZO-1/ZONAB信号通路的关系研究也是采用了RNAi技术[11]。由于RNAi技术存在不彻底性和时限性等缺点,因此,本研究通过CRISPR/Cas9基因编辑技术构建NPHP1基因稳定敲除的MDCK细胞模型,将有利于NPHP发病机制的体外研究。
CRISPR/Cas9技术是近年来发展较快的简便高效基因编辑技术[10,12],因操作简单易行,目前已广泛应用于基因功能研究[13]。该技术首先设计针对靶基因的sgRNA,通过构建重组质粒转染,使引导序列sgRNA和Cas9蛋白在细胞内表达,sgRNA引导Cas9蛋白至靶基因DNA的目标位置,对双链DNA进行切割。细胞通过NHEJ修复时,靶基因DNA目标区域可出现插入、缺失等突变。
本研究针对犬NPHP1基因第5外显子设计了3对sgRNA,该位点是NPHP1基因的主要功能结构域SH3结构域,是NPHP1编码蛋白nephrocystin-1与其他蛋白相互结合的重要位点[14]。nephrocystin-1通过SH3功能结构域与黏附连接相关的酪氨酸激酶配体p130Cas蛋白结合,共表达于黏附连接处,参与了调节细胞黏附连接和细胞骨架组装的信号通路[15]。Benzing等[16]的研究还表明,nephrocystin-1通过SH3功能结构域与p130Cas蛋白、张力蛋白tensin、富含脯氨酸的酪氨酸激酶2(Pyk2)等在细胞基底侧黏附连接处形成蛋白复合体,在肾小管上皮细胞的细胞连接方面发挥重要作用,nephrocystin-1可以调节Pyk2的402位酪氨酸磷酸化,并随后激活有丝分裂原活化蛋白激酶,参与了Pyk2相关信号通路。nephrocystin-1还可通过SH3功能结构域与PALS1结合调控Par3/Par6/aPKC复合体的Par6磷酸化状态,促进紧密连接形成[17]。
本研究通过CRISPR/Cas9技术成功构建了NPHP1基因稳定敲除的MDCK细胞。对靶基因目标位置的Sanger测序结果显示,同源DNA双链均发生了非3N移码突变,基因型为小片段缺失突变和点缺失突变的复合杂合突变:c.500_551del (p.His167GlnfsTer14) 和c.499delC (p.His167ThrfsTer31)。这两个突变均使NPHP1的氨基酸序列发生了移码突变,并使终止密码子提前出现,破坏了NPHP1基因的SH3功能结构域,使nephrocystin-1蛋白不再表达。同时,通过对本研究引导序列可能发生脱靶的8个位点进行Sanger测序检测,结果显示预测脱靶位点均未发现突变,基本排除了本研究构建的NPHP1敲除MDCK细胞的脱靶可能。RT-qPCR和Western blotting检测显示,NPHP1表达的mRNA水平显著受到抑制,编码蛋白nephrocystin-1蛋白表达完全缺失。NPHP1基因敲除MDCK细胞的细胞形态发生了改变,与通过慢病毒转染shRNA敲低NPHP1表达的MDCK细胞结果一致,略呈长梭形,细胞间隙稍增大。由此可见,本研究通过CRISPR/Cas9基因编辑技术成功构建了NPHP1基因稳定敲除的MDCK细胞,这将进一步促进NPHP发病机制的体外研究。
猜你喜欢
分子催化(2022年1期)2022-11-02
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年5期)2021-07-22
烟草科技(2021年6期)2021-06-24
江西农业学报(2021年4期)2021-04-20
科学导报(2021年3期)2021-02-22
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
