
江西地区24例罕见β-地中海贫血患者基因型分析
2020-06-29 07:43罗海艳刘艳秋谢康邹永毅陆清
实验与检验医学 2020年3期
罗海艳,刘艳秋,谢康,邹永毅,陆清
(江西省妇幼保健院产前诊断中心,江西 南昌 330000)
珠蛋白生成障碍性贫血(也称地中海贫血),是广泛分布于全球且危害性极大的血液系统常染色体隐性遗传性疾病,在中国主要高发于南部省份如广东、广西、海南等[1]。根据合成受到抑制的相应珠蛋白肽链类型,地中海贫血可分为α-地中海贫血、β-地中海贫血、δ-地中海贫血、γ-地中海贫血、δβ-地中海贫血、εγδβ-地中海贫血[2]。其中最常见的是α-地中海贫血和β-地中海贫血两类,致病机制主要由于α-珠蛋白或β-珠蛋白基因发生缺陷,致使珠蛋白肽链合成不足,使形成血红蛋白的α-链/β-链比例失衡,从而导致溶血性贫血[3]。编码α-珠蛋白的基因位于16 号染色体16p13.3 区域,主要的基因突变类型为大片段缺失突变,约占α-地贫的95%,部分α-地贫由点突变导致,约占α-地贫的5%[4];编码 β-珠蛋白的基因位于 11 号染色体11p15.3 区域,主要突变类型为点突变。目前我国已经报道了48 种β-地中海贫血基因点突变,其中常见的17 种β-地贫基因点突变,CD41-42(HBB:c.124_127delTTCT)、CD43(HBB:c.130G>T)、IVSII-654(HBB:c.316-197C>T)、-28(HBB:c.-78A>G)、-29(HBB:c.-79A>G)、-30(HBB:c.-80T>C)、-32(HBB:c.-82C>A)、CD71-72(HBB:c.216_217insA)、βE (HBB:c.79G>A)、CD17 (HBB:c.52A>T)、CD31 (HBB:c.94delC)、CD14-15 (HBB:c.45_46insG)地贫致病基因缺陷已纳入常规地贫基因检测范围[5-7]。但β-珠蛋白基因上其它的罕见位点突变或未知突变等,需通过Sanger 测序进行检测。少数β-地贫可由基因大片段缺失所引起,目前我国已报道了5 种大片段缺失导致的β-地中海贫血,包括Gantonese 缺失、Yunnanese 缺失、Chinese 缺失、S.E.Asian 缺失和 Taiwanese 缺失。其中 Chinese 缺失即 β-中国型缺失地贫 Gγ+(Aγδβ)0,S.E.Asian 缺失即β-东南亚缺失地贫SEA-HPFH,Taiwanese 缺失即β-台湾型缺失地贫在人群中更为常见。β-中国型缺失地贫 Gγ+(Aγδβ)0缺失约 79Kb,缺失位置包括 Aγ、δ、和 β 基因,β-东南亚缺失地贫 SEAHPFH 缺失约27 Kb,缺失位置包含整个β 基因,而β-台湾型缺失在β 基因上缺失约1357bp[8,9]。目前针对β-珠蛋白基因大片段缺失主要采用跨越断裂点多聚酶链反应(Gap-PCR)技术或多重连接酶依赖探针扩增技术(MLPA)方法进行检测[10,11]。
临床上, 血液学表型可作为β-地中海贫血患者的筛查指标, 根据RBC 指标和血红蛋白分析结果可初步诊断不同类型的β-地中海贫血基因携带者。β-地贫基因携带者一般具有小细胞低色素性贫血(MCV<80fl、MCH<27pg)表型,伴随血红蛋白电泳 HbA2 值升高(HbA2>3.5%)。因此,满足上述血液学表型的患者可视为β-地贫高风险个体。在临床检测中, 常规β-地贫基因检测可检出绝大部分β-地贫,但仍有少部分β-地贫高危患者可能存在罕见或未知突变而不能通过常规基因检测方法检出,容易导致漏诊、误诊。因此,对常规基因型检测结果与表型不符的β-地贫高危患者行进一步β-珠蛋白基因分析相当重要,尤其对于夫妻已有一方携带β-地贫基因突变的夫妇, 如另一方也携带罕见或未知β-地贫基因突变,会导致生育重型β-地贫患儿的风险大大增加。在这种情况下,我们对β-地贫基因诊断的基本流程为:反向点杂交技术分析常见的β-地中海贫血点突变→Gap-PCR 技术检测 β-中国型缺失地贫 Gγ+(Aγδβ)0和 β-东南亚缺失地贫SEA-HPFH 及Taiwanese 缺失→β-珠蛋白基因Sanger 测序分析罕见点突变。本研究通过Gap-PCR 技术和Sanger 测序对β-珠蛋白基因罕见突变类型进行诊断,试图找到β-地贫高危人群的罕见地贫基因突变位点。
1 研究对象与方法
1.1 研究对象 分析 2017 年 5 月至 2019 年 5 月期间于江西省妇幼保健院行β-地贫基因检测的患者,对符合小细胞低色素性贫血特征,即MCV<80fL,MCH<27pg,血红蛋白电泳 HbA2>3.5%,而常规 β-地贫基因检测为阴性的24 例DNA 样本进行后续β-地贫基因分析。该研究符合人体试验伦理学标准,并得到了伦理委员会的批准。
1.2 研究方法 对β-基因上可能存在的大片段缺失,参照徐湘民《地中海贫血预防控制操作指南》一书,进行多重Gap-PCR 扩增,对东南亚型HPFH(简称 SEA-HPFH)、中国型 Gγ+(Aγδβ)0与 Taiwanese 缺失型进行检测,设置阳性对照和正常内对照,样本不同缺失基因型经琼脂糖电泳后产物片段大小不同。对β-基因上可能存在的罕见点突变与未知突变, 设计2 对引物, 分别扩增HBB 基因上游-100 至 IVS-Ⅱ-81 与 IVS-Ⅱ-70 至下游终止密码子后Poly-A 两段,并行Sanger 测序。测序产物与HBB 基因参考序列比较,找出样本存在的罕见点突变与未知突变。具体操作步骤如下:
1.2.1 DNA 提取 在获取病人知情同意的前提下,采用天隆核酸提取试剂盒提取200μl 全血DNA,得到 90μl DNA 样品,调 DNA 浓度为 50~100ng/μl之间备用。
1.2.2 PCR 扩增 以提取好的DNA 样品为模板,加入 10×PCR Buffer、Taq 聚合酶、2.5mmol/L dNTPs、MgCl2、ddH2O 及 β-珠蛋白基因引物 (由上海生工公司合成),配制好缺失型突变及点突变检测反应体系, 在ABI 7300 PCR 扩增仪上扩增,β-珠蛋白基因缺失检测引物序列及扩增程序参照 《地中海贫血预防控制操作指南》一书。β-珠蛋白基因点突变检测引物序列见表1, 扩增程序为95℃预变性5min;95℃ 45s →62℃ 60s →72℃ 60s,35 个 循 环数;72℃延伸 5min。
1.2.3 产物分析 对进行β-珠蛋白基因大片段缺失型检测的PCR 产物行琼脂糖凝胶电泳, 分析是否存在相关缺失型突变;对进行点突变检测的PCR 产物行 Sanger 测序,与 β-珠蛋白基因 HBB 参考序列比对, 分析是否存在β-珠蛋白基因罕见突变与未知点突变。
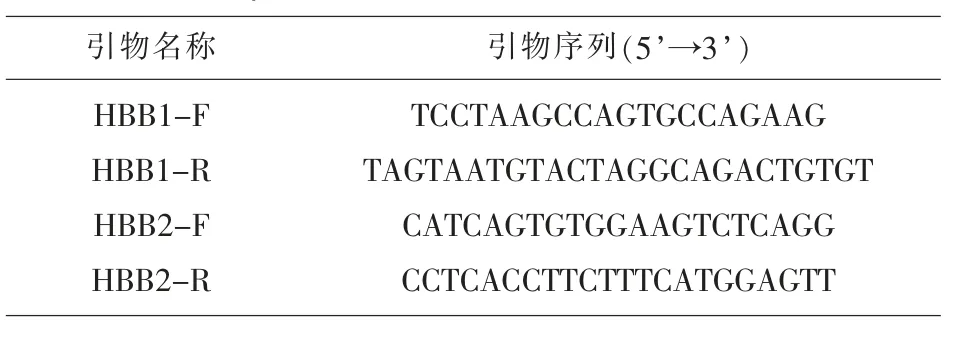
表1 β- 珠蛋白基因点突变检测引物序列
1.2.4 结果分析 电泳结束后,将样本条带与阴阳性对照进行比较,判断其缺失基因型类型。测序完成后,将测序图谱与HBB 标准参考序列进行比对,找出基因突变位点, 进入http://globin.bx.psu.edu/cgi-bin/hbvar 数据库进行查询,确定基因突变是否为报道过的已知致病突变或未知突变。
2 研究结果
通过对24 例疑似罕见β-地贫基因患者行Gap-PCR 及Sanger 测序分析,发现24 例患者均存在罕见突变,共计9 种突变类型,突变类型及突变人次如表2 所示。其中缺失型4 例, 点突变型20例, 典型β-珠蛋白基因缺失型突变电泳图如图1所示, 典型β-珠蛋白基因点突变测序图如图2 所示。

表2 9 种罕见β- 地贫突变类型
3 讨论
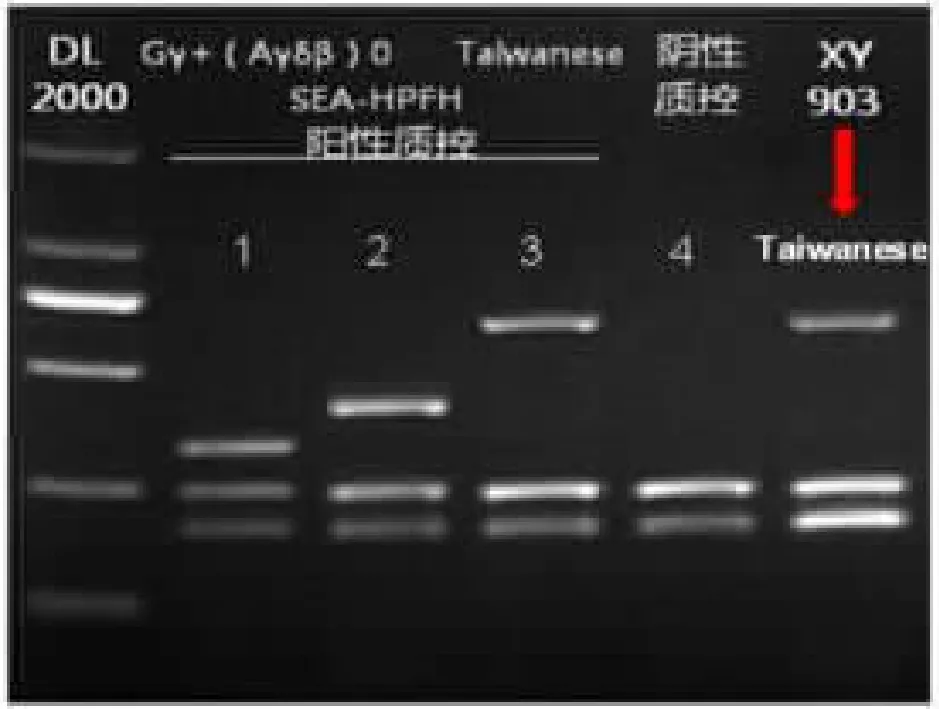
图1 典型缺失型罕见β- 地贫琼脂糖凝胶电泳示意图
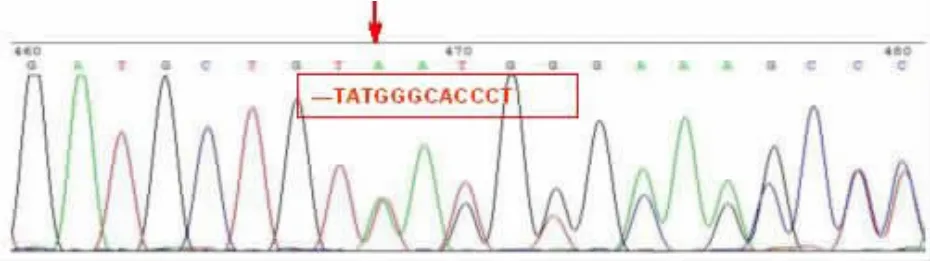
图2 典型点突变型罕见β- 地贫Sanger 测序图
β-珠蛋白基因突变导致的重型β-地贫危害极大,患儿出生时不会表现出疾病类型,通常在出生后几个月或一周岁内开始出现慢性进行性贫血症状[12]。患者除严重贫血外,易伴有轻度黄疸,肝脾肿大,发育不良,并具有典型的地中海贫血面容,如头大、眉距增宽、鼻梁低平、颧骨突出等。常并发支气管炎或肺炎,当并发有含铁血黄素沉着时,因过多的铁沉着引起心脏、肝、胰腺、脑垂体等脏器的损害,严重时可导致死[13]。重型β-地贫患儿如不加以治疗,多于5 岁之前死亡。目前,重型β-地贫尚无理想的治疗手段,主要通过长期定期输血并通过铁螯合剂进行祛铁治疗,治疗繁琐且费用昂贵,一般情况下患儿和家长都难以接受。造血干细胞移植(HSCT)是目前能根治重型β-地贫的方法,根据干细胞来源可分为骨髓移植、外周血干细胞移植和脐血移植。患儿尽早接受HSCT,移植效果越好,但该方法存在HLA 配型相符的造血干细胞供体来源受限的缺点[14-16]。因此,通过人群筛查发现常见及罕见的β-地贫携带者, 并对于携带同型地贫基因的育龄夫妇提供产前诊断与遗传咨询,有助于合理指导妊娠,降低出生缺陷。
分析结果表明,24 例存在罕见β-地贫突变的样本中, 包括3 种内含子区突变,4 种外显子区突变,2 种缺失型突变,所有突变基因型均与其临床表型相符。可以看出导致β-地贫的基因突变分布区域较广,在编码区和非编码区均有出现。在本次研究中有一些突变位点出现频率较高,例如:CD54-58(-TATGGGCACCCT) β0杂合突变共计 8 例,-31(A>G) β+杂合突变共计 3 例。所分析样本中,检出 2例β-地贫复合杂合突变,即IVS-II-81(C>T)复合-28 (A>C) β+突变,查询 HbVar 数据库提示 IVS-II-81(C>T)突变可能为中性多态性。对这两例患者进行家系分析,发现第一名患者的IVS-II-81(C>T)突变遗传自母亲,其母亲血液学表型未见异常;第二名患者的IVS-II-81(C>T)突变遗传自父亲,其父亲血液学表型也未见异常,这更进一步提示IVS-II-81(C>T)可能为人群中性多态性变异。同时我们发现IVS-I-6(T→G)突变在HbVar 数据库未见报道,但有文献报道表明当该位点突变成C 碱基IVS-I-6(T→C)时可导致 β+地中海贫血[17],因此我们推测IVS-I-6(T→G)也很有可能与该患者的地中海贫血表型相关。
在临床实践工作中,对于表型与基因型不符的患者,应行罕见β-地贫基因检测以明确其基因型,防止误诊或漏诊。下列情况应高度怀疑为罕见β-地贫患者:⑴呈小细胞低色素贫血 (MCV<80fL,MCH<27pg)且血红蛋白电泳提示HbA2>3.5%而常规基因检测未见异常的患者;⑵呈中、重型β-地贫表型,但常规基因型检测仅为携带者的患者。因此在常规17 种β-珠蛋白基因突变检测的基础上,结合多种分子方法对β-地贫高危患者行罕见β-地贫突变分析,可使β-地贫基因检测更加准确,发现罕见突变或新突变,对指导地贫基因产前诊断、降低出生缺陷具有重要作用。
猜你喜欢
传染病信息(2022年4期)2022-11-23
西部医学(2022年9期)2022-09-26
中国现代医生(2022年21期)2022-08-22
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
生物学教学(2018年8期)2018-09-03
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
