
聚乙烯醇 -十二烷基硫酸钠 -Fe(OH)3微行为研究
2020-06-21 11:50:06程宏英夏嗣禹周宇杰董延茂
苏州科技大学学报(自然科学版) 2020年2期
程宏英,夏嗣禹,周宇杰,魏 杰,董延茂
(苏州科技大学 化学生物与材料工程学院,江苏 苏州215009)
Fe(OH)3溶胶粒子呈棒状或杆状,在结构、形状和粒径的研究中,常借助聚合物或氨基酸网格介质对其进行固定[1-2]。固定作用的强弱主要取决于待测分子与聚合物之间作用力的大小[3]。对于在聚合体系中易发生团聚或因团聚而失活的胶体,这种网格固定作用无法有效控制胶粒的有序分散,而使得测定结果产生误差[4]。选择能有效分散杆状Fe(OH)3胶粒的聚合体系对于研究胶体的结构和性质有其积极的作用。
Fe(OH)3溶胶制备最常用的方法是FeCl3溶液在沸水中水解后进行渗析而得。但是,不同的条件制备所得的胶体粒径大小不一,且由于Fe(OH)3溶胶胶团扩散层是高度分散的水化层,以及棒状的溶胶粒子在溶剂(水)中无序的排列,因此,纳米粒度测定仪无法直接测定Fe(OH)3溶胶的粒径。
实验探讨了Fe(OH)3溶胶制备过程中FeCl3溶液的浓度、溶胶的渗析时间和搅拌速度对其粒径的影响,运用傅里叶红外(FTIR)研究了改性阴离子复合体系PVA-SDS 与Fe(OH)3溶胶之间的性质影响和相互作用,发现Fe(OH)3溶胶在PVA-SDS 阴离子复合体系中仍保留丁达尔效应的性质,为Fe(OH)3溶胶在聚合物体系中的应用打下基础。
1 实验部分
1.1 仪器与试剂
UV-2550 紫外光度仪(日本,岛津),HH-1 型恒温水浴锅(金坛新航),JJ-1 机械电动搅拌器(浙江,金华),傅里叶红外(FTIR)(美国珀恩),S-4700 扫描透镜(SEM)(日本,岛津),纳米粒度及ZETA 电位分析仪(美国,Microtrac Inc),照度计(日本,EKO),冷冻干燥机(江苏天翔)。
十二烷基硫酸钠(SDS)(AR,上海凌风化学试剂),聚乙烯醇(PVA)(AR,苏州东都化学试剂),其他常用试剂均为分析纯,水为蒸馏水。
1.2 渗析法处理氢氧化铁溶胶
1.2.1 水解法制备Fe(OH)3溶胶
准确称取10.0 g FeCl3·6H2O 溶于90 mL 蒸馏水中。分别移取1、3、5、7、9、11 mL 的上述溶液在搅拌情况下滴入200 mL 100 ℃的水中(控制在3-5 min 内滴完),水解过程中CFeCl3分别为0.002、0.006、0.010、0.014、0.018、0.022 mol·L-1。继续煮沸2 min,即得渗析前Fe(OH)3溶胶[5]。
1.2.2 Fe(OH)3溶胶渗析
待上述样品冷却至30 ℃左右,分别装入预先准备好的玻璃纸袋中,将袋口系好浸入蒸馏水中进行渗析。每隔1 h 换水1 次。为研究渗析时间对胶体的行为影响,渗析时间分别在1、2、3 天时进行取样胶体。
1.3 Fe(OH)3 溶胶的行为研究
1.3.1 胶体反射率的测定
选择反射率作为测定数据,在仪器调零和基线扫描后,将待测样品分别进行反射率和反射波长的扫描。
1.3.2 胶体粒径的测定
用纳米粒度及Zeta 电位分析仪分别测定PVA-SDS 以及PVA-SDS-Fe(OH)3溶胶体系的粒径,运用仪器配套软件求得被测样品的平均粒径。
1.3.3 胶体丁达尔效应研究
将一束红光依次透过Fe(OH)3溶胶、PVA-SDS-Fe(OH)3体系和PVA-SDS 体系,观察溶液中是否产生一条光亮的通路,并运用光照计测量通路的光强。
2 结果与讨论
2.1 FeCl3 用量与渗析时间对Fe(OH)3 溶胶反射能力的影响
胶体的粒径大小与形态对于光在其表面发生反射、折射或衍射能力有直接的影响[6]。图1 为FeCl3用量、溶胶渗析时间和搅拌速度对胶体紫外-可见光谱反射的影响。由图1A 可知,随着FeCl3用量从1 mL 到11 mL 的增加,溶胶反射率逐渐下降,反射波长逐渐红移。而图1B 显示随着渗析时间从0-3 d 的延长,反射率逐渐下降,且反射波长缓慢红移。图1C 显示制备过程中搅拌速度从60 r·min-1增加到160 r·min-1,Fe(OH)3溶胶反射波长逐渐蓝移。图1 表明,FeCl3用量、溶胶渗析时间和搅拌速度对Fe(OH)3溶胶胶粒的粒径有比较大的影响。
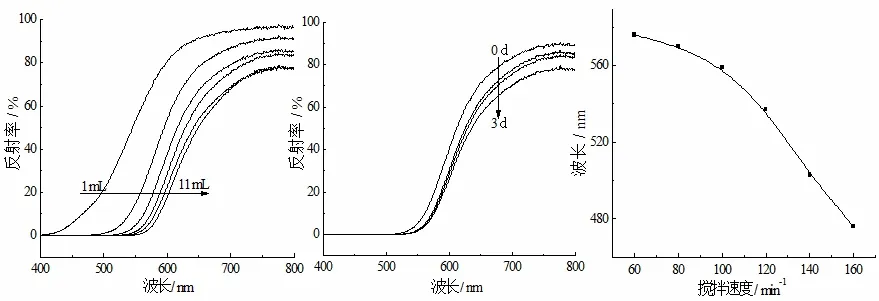
图1 FeCl3 用量(A)、渗析时间(B)和搅拌速度(C)对制备所得溶胶紫外-可见反射光谱的影响
2.2 PVA-SDS 和PVA-SDS-Fe(OH)3 体系粒径的测定
由文献[7]可知,Fe(OH)3溶胶胶粒呈杆状,高度分散的水化层不利于胶粒粒径直接响应。因此,选择相反电荷的聚合物对Fe(OH)3溶胶胶粒进行固定,是研究其形状和结构常用的方法。实验配置50 mL 10%(w/w)的SDS 溶液,60 ℃下恒温30 min,加入0.5 g PVA 超声30 min 后加热至全部溶化,80 ℃下继续恒温30 min,即得复合改性阴离子体系(PVA-SDS)。冷至室温后,加入适量的待测Fe(OH)3溶胶,手摇混匀在Zeta纳米粒度测定仪上测定其平均粒径并与PVA-SDS 体系进行比较。图2 是FeCl3浓度对PVA-SDS-Fe(OH)3复合体系粒径的影响,其中插图是PVA-SDS 粒径的分布图。由图可知,实验测得PVA-SDS-Fe(OH)3体系的平均粒径随着FeCl3浓度的增加而逐渐增大, 在0.008-0.012 mol·L-1区间内平缓上升,表明在此浓度范围内,Fe(OH)3溶胶体系的粒径缓慢而均匀增长,溶胶稳定性高。PVA-SDS 体系的平均粒径比PVA-SDSFe(OH)3的体系大1-2 个数量级。表明PVA-SDS 与Fe(OH)3胶粒间的作用力改变其结合物的结构,从而降低了体系的平均粒径。
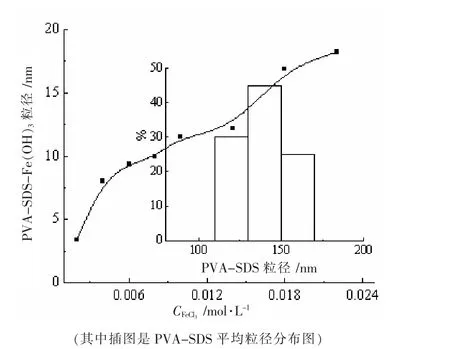
图2 FeCl3 浓度对PVA-SDS-Fe(OH)3 复合体系粒径的影响
2.3 Fe(OH)3 溶胶和PVA-SDS-Fe(OH)3 复合体系胶体性质的研究
由于Fe(OH)3溶胶的胶粒直径小于入射光的波长发生散射形成光的通路即为丁达尔效应,可用于区分胶体与溶液[8]。通路的强度,随着胶粒半径增加而发生变化。实验将Fe(OH)3溶胶、PVA-SDS-Fe(OH)3复合体系以及PVA-SDS 三份样品依次排列,取一束光源透过三份体系,从垂直于入射光的方向观察丁达尔效应。由图3 可知,PVA-SDS 体系中没有产生光亮通路,即无丁达尔效应,表明PVA-SDS 体系不是胶体。而在Fe(OH)3溶胶和PVA-SDS-Fe(OH)3复合体系中均有一条明显的光亮通路。其中,PVA-SDS-Fe(OH)3复合体系中的光通路强度明显比Fe(OH)3溶胶体系强,表明PVA-SDS-Fe(OH)3复合体系仍保留了Fe(OH)3胶体的性质,且因体系粒径发生变化,因而增强了丁达尔效应的光亮强度。
同时,实验考察了PVA(1%,w/w)-SDS(10%,w/w)改性阴离子聚合物中,胶体Fe(OH)3的浓度对复合体系光亮通路的影响,采用光照计测量丁达尔效应的强度值见图4。在PVA-SDS 体系中,随着胶体浓度的增大,丁达尔效应的强度逐渐增强,在1.0-2.0 mmol·L-1浓度范围内,增加幅度逐渐平缓,在1.5-2.0 mmol·L-1范围内强度达到峰值,表明PVA-SDS 与胶体Fe(OH)3之间的单分子作用达到最大。随着Fe(OH)3胶体浓度继续增大,过多的正电荷胶粒在PVA-SDS 体系中逐渐团聚,丁达尔效应强度逐渐下降。

图3 Fe(OH)3 溶胶、PVA-SDS-Fe(OH)3 体系以及PVA-SDS 改性聚合物的丁达尔效应比较
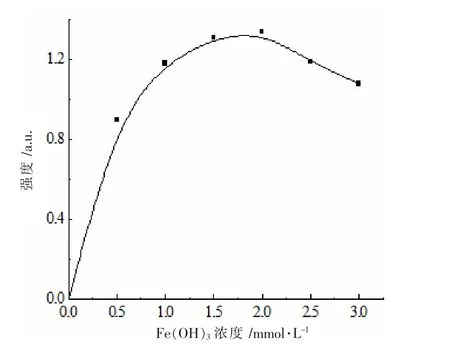
图4 PVA-SDS-Fe(OH)3 不同浓度复合体系对丁达尔效应强度的影响
2.4 PVA-SDS 与Fe(OH)3 溶胶作用机理研究
实验采用FTIR 技术表征PVA-SDS 体系与PVA-SDS-Fe(OH)3复合体系的作用机理,如图5 所示。在PVA-SDS 红外图谱中,1048 cm-1为-SO3Na 基团的对称伸缩振动吸收峰,1221 cm-1为-SO3Na 基团的反对称伸缩振动吸收峰。而在PVA-SDS-Fe(OH)3红外图谱中,-SO3Na 基团的对称伸缩振动峰从1048 cm-1蓝移到了1030 cm-1,且振动强度与对应位置的反对称伸缩振动都明显减弱。这表明,PVA-SDS 改性阴离子聚合物与带正电荷的Fe(OH)3胶粒之间发生了静电作用,由于Fe(OH)3胶粒中心正电荷的电子云密度转移而使得-SO3Na 基团的伸缩振动发生相应的改变[9]。
在PVA-SDS 体系中,由于相似相溶原理和极性头基之间的相互排斥作用,PVA-SDS 呈球簇形以提高其稳定性[10]。Fe(OH)3胶体在水解和渗析过程中,形成了非常稳定的{[Fe(OH)3]m·nFeO+·(n-x)Cl-}x+·xCl-胶团结构,紧密层与扩散层之间的电位差使得胶粒外层的水化层均一稳定的分散在其周围,因此杆状的Fe(OH)3胶体在水中呈高度分散的状态。而在PVA-SDSFe(OH)3复合体系中,由于静电作用,PVA-SDS 与Fe(OH)3胶粒的排列由团簇而变成线,如图6 所示,从而使PVA-SDS-Fe(OH)3复合体系的平均粒径比PVA-SDS 小近1-2 个数量级。
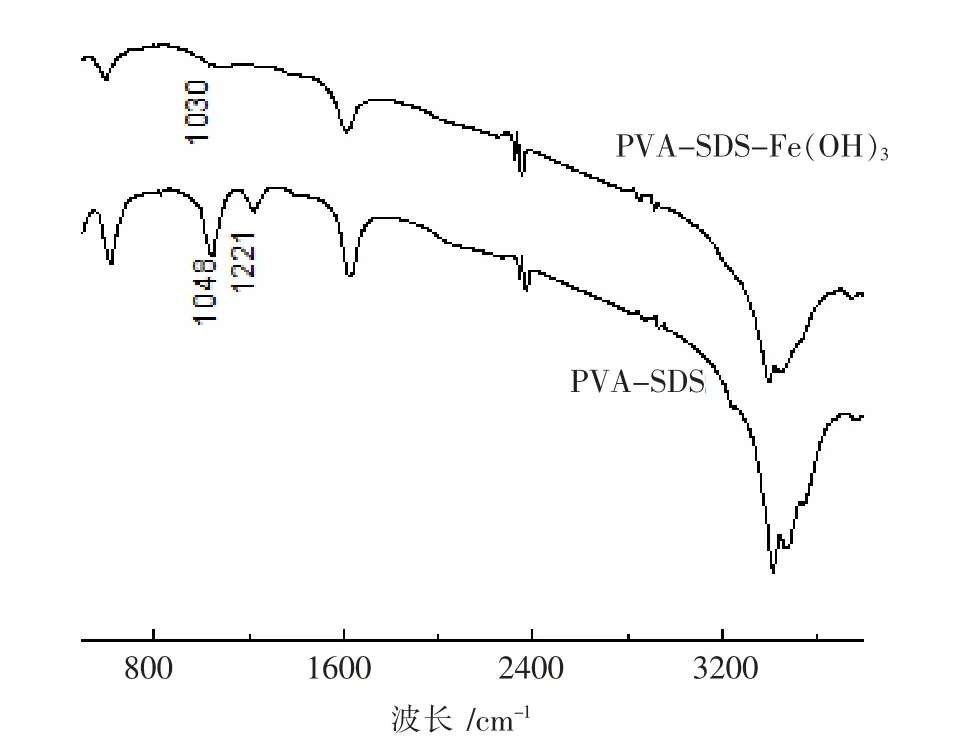
图5 PVA-SDS 与PVA-SDS-Fe(OH)3 体系的红外图谱
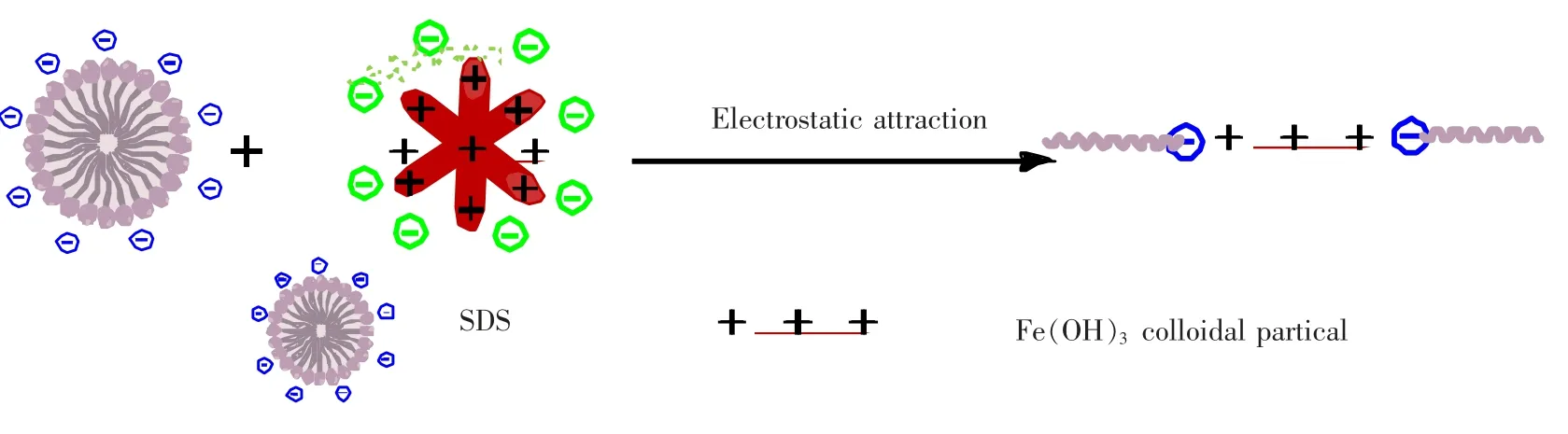
图6 PVA-SDS 与Fe(OH)3 溶胶作用机理示意图
另外,实验采用SEM 对PVA-SDS-Fe(OH)3进行形状表征,如图7 所示,PVA-SDS-Fe(OH)3体系冷冻干燥后的扫描透镜图见图7(A),方框内局部区域放大10 倍图见图7(B)。由图7(A)可知,经过迅速冷冻干燥处理后,PVA-SDS 团聚成簇状,而Fe(OH)3胶体呈线状,由于异性电荷间的相互作用,Fe(OH)3胶粒分散在PVA-SDS 周围。从其放大图7(B)可知,Fe(OH)3胶体长度达1-2 μm,粒径10-20 nm。
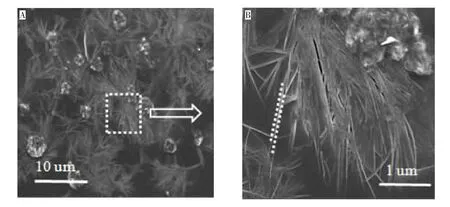
图7 PVA-SDS-Fe(OH)3 的SEM 图
3 结语
Fe(OH)3胶体的粒径与制备过程中FeCl3的用量、搅拌速度和胶体的渗析时间有相关影响,改性阴离子复合体系PVA-SDS-Fe(OH)3保留胶体的性质,且由于异性电荷的非共价吸引,PVA-SDS 与Fe(OH)3胶体的排列由团簇变成线状,提高了Fe(OH)3胶体在水相中的稳定性,为Fe(OH)3胶体在不同环境中的应用条件研究打下一定的基础。
猜你喜欢
知识就是力量(2022年11期)2022-05-30 06:00:05
石油沥青(2022年2期)2022-05-23 13:02:36
发明与创新(2021年44期)2021-12-15 10:27:10
腐植酸(2021年2期)2021-12-04 04:27:17
环球人物(2021年18期)2021-09-28 22:48:27
意林原创版(2021年12期)2021-01-16 02:58:02
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
四川水泥(2018年10期)2018-10-24 07:36:20
东北水利水电(2017年8期)2017-08-17 10:58:40
中国农村水利水电(2016年7期)2016-03-22 06:55:03
