
RRM2B基因c.420G>C新突变细胞模型的建立及初步研究
2020-06-11 06:06:12王建力畅雪丽郭军红
中西医结合心脑血管病杂志 2020年9期
王建力,畅雪丽,张 炜,郭军红
线粒体神经胃肠脑肌病(mitochondrial neurogastrointestinal encephalomyopathy,MNGIE)是一组常染色体隐性遗传的罕见病,临床表现包括眼睑下垂和/或眼外肌麻痹、胃肠蠕动障碍、周围神经病变;肌肉病理检查可发现破碎红纤维(ragged-red fibers,RRFs)和破碎蓝纤维(ragged-blue fibers,RBFs),头颅磁共振成像(MRI)可见广泛的脑白质病变[1-3]。目前认为其致病基因为位于常染色体22q13.32的TYMP基因,该基因编码胸苷磷酸化酶(thymidine phosphorylase,TP)。TYMP基因突变致TP功能异常,使脱氧胸苷和脱氧尿苷大量蓄积于组织器官中,从而导致三磷酸脱氧核苷酸库(dNTPs)的平衡被破坏,引起病人一系列临床表现。
RRM2B基因位于常染色体8q22.3,编码p53-诱导型核糖核苷酸还原酶(ribonucleotide reductase,RNR)的小亚基(p53inducible small subunit of the ribonucleotide reductase,p53R2)。RNR可以催化核糖核苷酸(nucleoside diphospate,NDP)还原生成相应脱氧核糖核苷酸(deoxy-ribonucleotide diphosphate,dNDP),是体内三磷酸脱氧核糖核苷酸(deoxy-nucleoside triphosphate,dNTP)从头合成的限速酶和关键酶[4]。p53R2对于dNTPs的合成十分重要,对于需要不断复制的线粒体脱氧核糖核酸(mitochondrial DNA,mtDNA)更是如此;RRM2B基因突变导致dNTP从头合成途径异常,从而导致mtDNA耗竭综合征(mtDNA-depletion syndromes,MDS)[5]。MDS是一组mtDNA拷贝数严重减少导致受影响组织和器官的能量生产受损的常染色体隐性遗传疾病[6]。RRM2B基因突变所致的MDS一般表现为癫痫、肌病、胃肠道功能紊乱、乳酸酸中毒、肾小管肾病,甚至幼年死亡[7]。然而MDS仍有较为轻微的临床表型报道,病人甚至成年起病,临床表现为MNGIE[8]。
本研究报道1例临床主要表现为脑白质病变、呼吸衰竭、听力下降、眼外肌麻痹、腹泻及恶液质的中年女性病人,临床拟诊MNGIE,基因检测结果提示RRM2B基因c.420G>C纯合突变。
1 方 法
1.1 临床数据收集 超过3位专业的神经科医生确定病人临床表现,收集病人生化指标及影像学结果;股四头肌处进行肌肉活体组织检查;留取病人静脉血,康旭医学检测中心对37个线粒体基因和185个线粒体相关核基因进行检测及数据分析。
1.2 细胞模型
1.2.1 细胞培养 37 ℃,5% CO2,DMEM细胞培养基添加10%胎牛血清培养293T细胞。
1.2.2 质粒转染 转染前24 h,在不含抗生素的完全培养基中接种细胞,细胞密度80%~90%时转染质粒。48 h后收集细胞。
1.2.3 Western Blot试验 裂解液提取细胞蛋白,12 000 r/min离心10 min,97 ℃加热10 min后进行实验。蛋白经SDS-PAGE胶分离后转移至NC膜;5%脱脂奶粉封闭1 h,在4 ℃下用p53R2/GAPDH抗体孵育过夜。洗膜后,二抗4 ℃孵育过夜。洗膜并检测结合抗体。
1.2.4 mtDNA含量检测 提取细胞(5×106)的线粒体DNA,利用分光光度计测定线粒体DNA浓度。
1.3 文献检索 根据关键词RRM2B、线粒体在PubMed、万方数据库、中国知网进行检索。
1.3.1 纳入标准 ①公开发表的期刊论文、著作、会议论文及摘要、学位论文;②病人经基因检测发现明确的RRM2B基因变异;③文献中给出详细的临床表现,包括病人性别、年龄、临床表现、各临床表现出现的时间及生化指标和辅助检查。
1.3.2 排除标准 ①未行基因检测或经基因检测未能明确致病基因;②未对病人临床表现进行详细描述或临床表现以统计学形式给出;③重复发表的病例;④病人为杂合突变,临床表现极为轻微或仅在特殊诱因下发病。
2 结 果
2.1 临床资料
2.1.1 临床表现 1例40岁女性病人,由于腹泻及体重减轻入住我院。病人足月顺产,6岁使用庆大霉素后双耳听力受损,之后听力逐渐下降,至37岁听力完全丧失;7岁时曾诊断为过敏性紫癜(具体不详);27岁因外伤行头颅CT检查,提示脑白质病变;31岁时病人出现进行性肢体无力,活动后加重;33岁时因发热诊断为慢性肾炎(具体不详);37岁开始无明显诱因出现排便次数增多,每日4次或5次,成形;37~40岁逐渐出现吞咽困难、眼睑下垂、视物成双,体重减轻共25 kg。家族史:祖母与外祖母系同村同姓人家,具体亲缘关系不详;其父排便次数多,每日4次或5次,成形;32岁剖宫产一女,其女体健。病人身高158 cm,体重34 kg(体质指数13.62 kg/m2)。神经系统查体:高级智能活动正常;双侧瞳孔等大等圆,直径约4.0 mm,对光反射存在;双侧眼睑下垂,眼球活动差;双耳听力完全丧失;屈颈无力,吞咽困难,双侧软腭上抬无力,四肢近端肌力差,四肢腱反射减弱;四肢末梢浅感觉减退,余神经系统查体大致正常。
2.1.2 生化指标 尿糖轻微增高;血常规、尿常规、凝血、肝肾功能、电解质、血糖、肌酶、甲状腺功能、风湿系列、血浆乳酸水平大致正常。
2.1.3 影像学检查 头颅MRI提示弥漫脑白质病变(见图1),磁共振波谱分析(MRS)未见乳酸峰增高。肌电图及神经电图提示轻度神经源性损害。上消化道中消化道造影提示:慢性胃炎、胃下垂;肠功能紊乱(通过过快);横结肠低位。
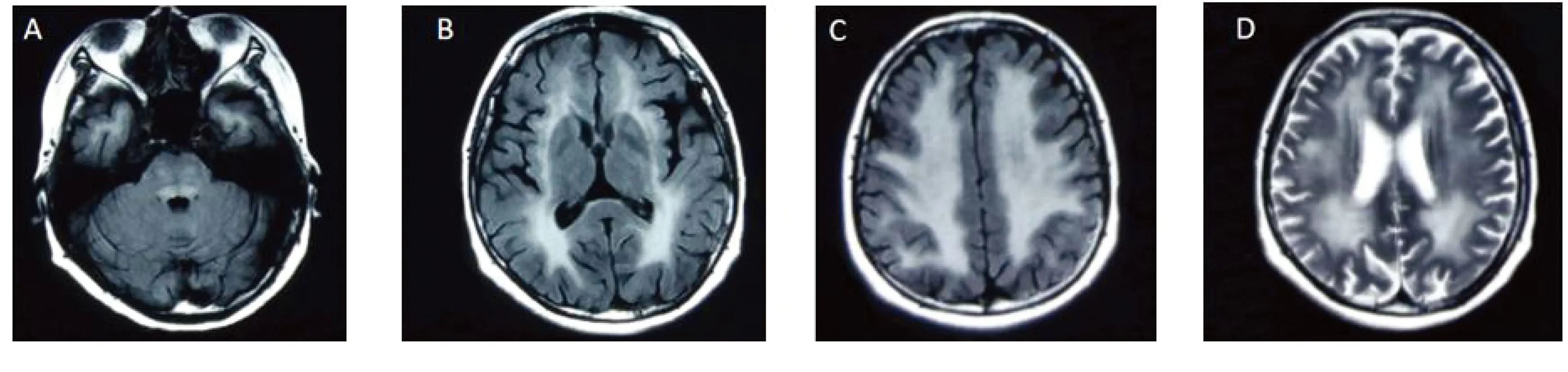
图1 头颅MRI提示脑白质病变
2.1.4 肌肉病理检查 征得病人家属同意后,对病人进行肌肉病理检查。结果提示:肌纤维大小重度不等,小圆变肌纤维散在存在,苏木精-伊红(HE)及改良格莫瑞三色(MGT)染色可见RRFs;琥珀酸脱氢酶(SDH)染色可见大量RBFs,细胞色素氧化酶(COX)染色可见部分肌纤维COX酶活性缺失:ATPase染色提示Ⅱ型纤维萎缩;油红 O 染色液(ORO)染色及过碘酸雪夫氏(PAS)染色未见异常。详见图2。
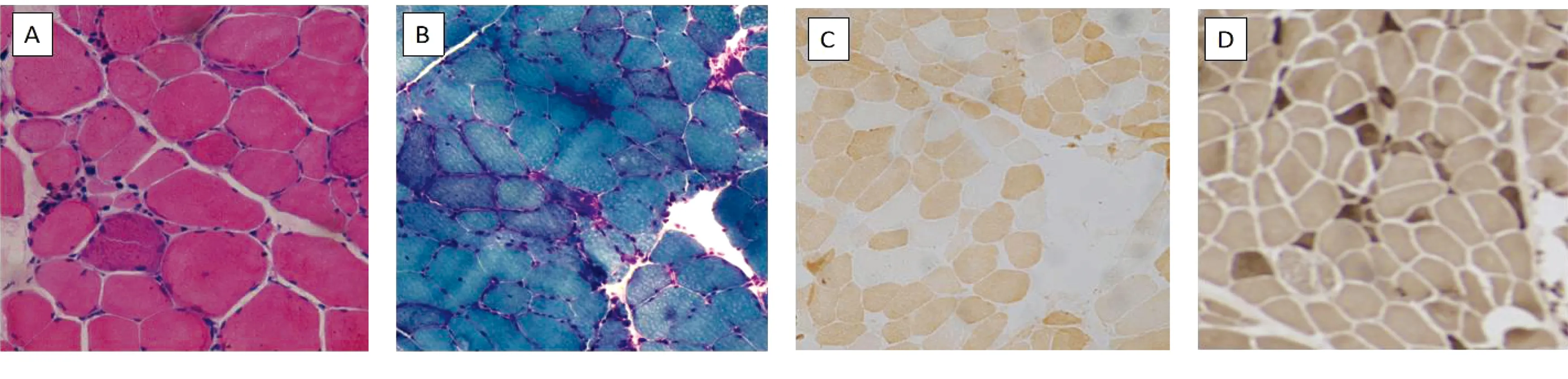
图2 肌肉病理检查可见RRFs(×100)
2.1.5 基因检测 在征得病人家属同意后对病人进行基因检测。检测线粒体基因组上的37个基因,未发现与临床表型相关的基因变异,进一步检测未发现病人线粒体基因大片段变异;检测线粒体病相关185个核基因,在病人RRM2B基因发现c420.G>C(p.Leu140Phe)的纯合核苷酸变异,对病人父母该位点进行Sanger测序,证实其父母该位点均为杂合子(见图3)。该突变在ExACA、ClinVar及OMIM数据库均未见收录。
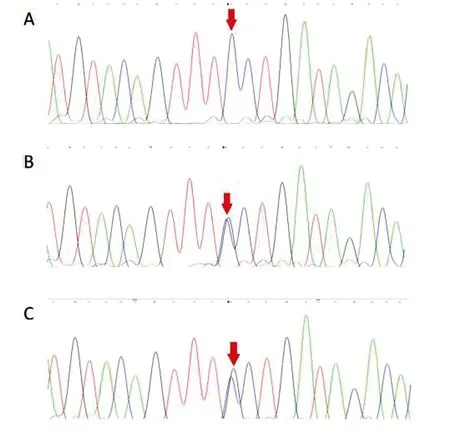
图3 基因检测结果(箭头指示突变位点)
2.1.6 随访结果 2年后,病人因发热导致呼吸衰竭再次入住我院。查体:体质指数较前无明显改变;右侧瞳孔3.5 mm,左侧瞳孔3.0 mm,对光反射消失;其他体征较前无明显改变。血气分析(鼻导管吸氧状态下):pH 7.09,动脉血氧分压103 mmHg(1 mmHg=0.133 kPa),动脉血二氧化碳分压108 mmHg。立即给予病人抗感染及呼吸支持治疗;在院期间反复发生严重感染,6个月后病人家属要求带管离院(气管切开状态),院外继续家用呼吸机辅助呼吸;规律随访1年后失访。
2.2 细胞模型 干扰质粒(pLVX-shRNA2-shR RM2B)转染293T细胞抑制293T细胞RRM2B基因转录,之后转染含突变的过表达质粒[Plvx-IRES-ZsGreen1-RRM2B(c.420G>C)]模拟RRM2B基因c.420G>C的突变细胞。
2.2.1 RRM2B基因c.420G>C突变降低293T细胞内RRM2B基因蛋白表达水平 RRM2B基因是编码p53R2蛋白的核基因。Western Blot法测定细胞内p53R2蛋白的数量(见图4),在含突变细胞内p53R2蛋白含量显著下降,即RRM2B基因蛋白表达量下降。同时,在转染过表达质粒(pLVX-res-ZsGreen1-RRM2B)后可以逆转这种下降趋势。
2.2.2 RRM2B基因c.420G>C突变降低293T细胞内mtDNA含量 p53R2蛋白对于mtDNA的生成十分重要,所以推测RRM2B基因c.420G>C突变同样可以影响mtDNA数量并导致MDS。提取细胞内mtDNA,利用分光光度计测定mtDNA含量(见图5)。在突变细胞内mtDNA数量显著下降,同时,在转染过表达质粒(pLVX-res-ZsGreen1-RRM2B)后mtDNA含量恢复。

A:空白组,无任何处理;B:干扰质粒的空白对照;C:过表达质粒的空白对照;D:实验组,转染含点突变的过表达质粒+干扰质粒;E:对照组,转染不含点突变的过表达质粒+干扰质粒。与实验组比较,*P<0.001。图4 RRM2B基因c.420G>C下调基因表达量

A:空白组,无任何处理;B:干扰质粒的空白对照;C:过表达质粒的空白对照;D:实验组,转染含点突变的过表达质粒+干扰质粒;E:对照组,转染不含点突变的过表达质粒+干扰质粒。与实验组比较,*P<0.001。图5 RRM2B基因c.420G>C降低细胞内mtDNA含量
2.3 文献回顾 根据纳入标准,初次检索后得到76篇文献;根据排除标准最终得到9篇文献[7-15]。根据9篇文献,统计来自12个家系的13例RRM2B基因变异病人(包含本研究纳入的1例病人)的临床数据,总结RRM2B基因变异的临床特点。详见表1。
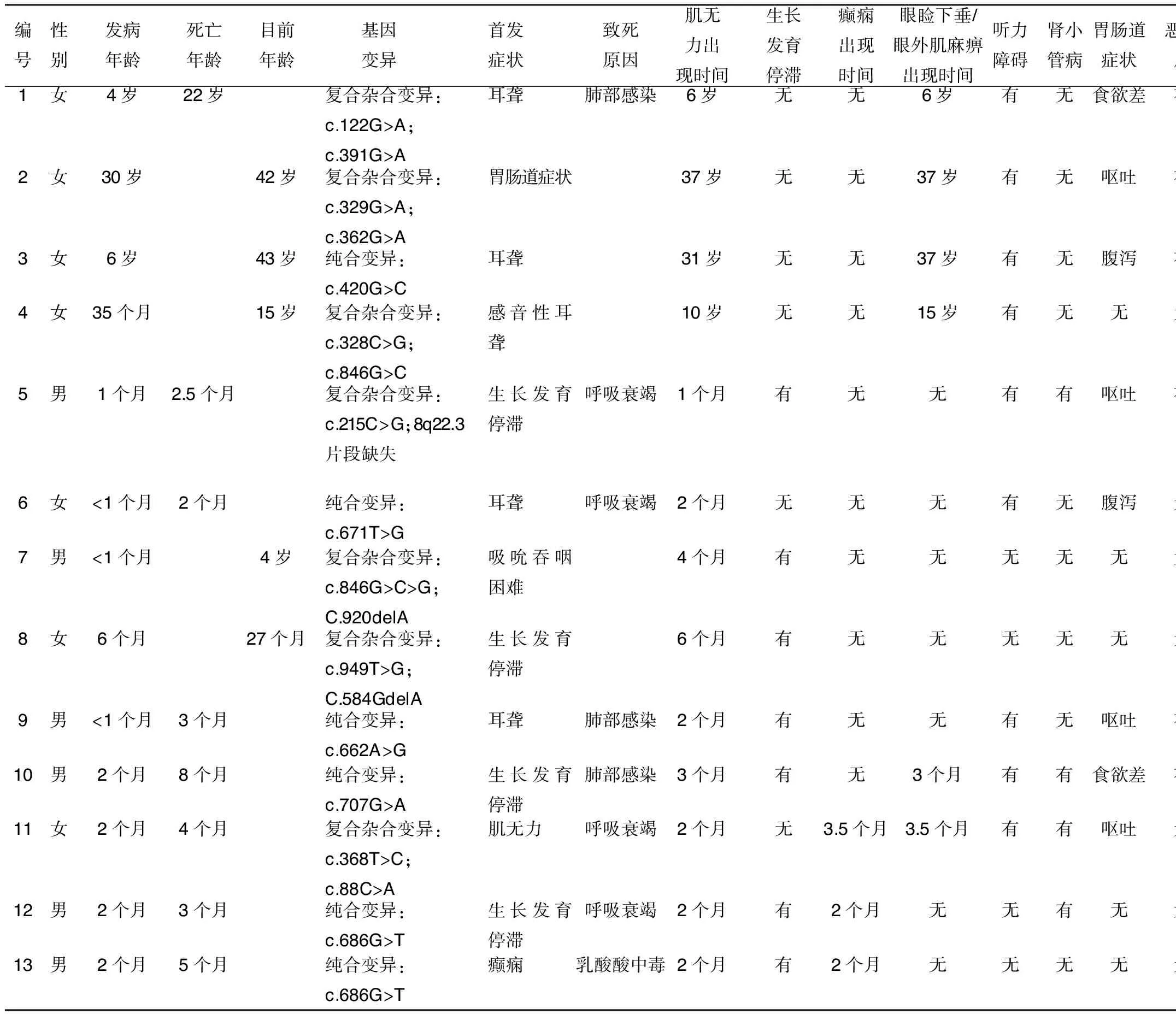
表1 13例RRM2B基因突变病人临床表现及辅助化验检查指标
3 讨 论
哺乳动物中,mtDNA共编码13种线粒体蛋白质;但是mtDNA的数量及表达依赖于数百个核基因编码的在细胞质中合成后输入线粒体的蛋白质,这些基因决定了mtDNA的数量、复制、转录、RNA成熟和翻译[16]。这一类核基因可以分为两类:一类是编码的蛋白可以直接作用于mtDNA复制叉的基因,例如POLG、POLG2和TWINKLE;一类是编码的蛋白可以为mtDNA复制提供所需的dNTPs,例如TK2、DGUOK、TYMP、SUCLA2、ANT1[17]。也就是说,这些核基因的变异可以影响线粒体内的核苷酸平衡或mtDNA复制进而导致mtDNA异常。
RRM2B基因是编码p53R2的核基因,位于8q22.3。两个拷贝数的p53R2与两个拷贝数的R1一起形成RNR;RNR可以催化NDP还原生成相应dNDP,为DNA的合成和修复提供前体[18]。p53R2对于非增殖细胞中DNA修复和mtDNA合成所需的dNTPs合成至关重要[19]。
RRM2B基因变异主要影响的器官是骨骼肌、大脑及肾脏[20]。RRM2B基因变异导致MDS,表现为癫痫、肌张力降低、腹泻、肾小管病变、乳酸酸中毒和婴幼儿早期死亡[7]。
缺乏p53R2会阻碍氧化损伤后DNA的修复并激活p53依赖的细胞凋亡。由于肾脏对于清除体内有毒代谢产物十分重要,意味着肾脏经常暴露于各种氧化应激环境下,因此RRM2B突变常导致严重的肾脏损害[21]。RRM2B基因突变所致疾病的临床表现可能存在组织器官特异性,这可能与胚胎早期细胞核DNA突变致mtDNA复制异常,在不同干细胞之间mtDNA损伤或耗竭的程度不同[22];随着干细胞的分裂分化产生了组织器官特异性,同样可以解释不同病人之间发病年龄、病情严重程度差异较大的原因。本研究所报道的1例病人临床表现为胃肠道功能紊乱、周围神经损害、RRFs和脑白质病变,完全符合MNGIE的临床特征,基因检测发现RRM2B基因c.420G>C纯合核苷酸变异。目前已有文献报道成年RRM2B基因突变病人临床表现为MNGIE[8]。事实上,RRM2B基因突变病人临床常伴随有胃肠道功能障碍,出现类似MNGIE病人临床表型,称为MNGIE-like[23]。
通过对所统计的文献中13例病人的临床表现、生化指标及辅助检查结果总结:①所有病人均出现不同程度的肌无力,且肌无力出现后常伴随着病人病情的严重暴发,提示肌无力出现的早晚可能与病人病程长短呈正相关,同时提示临床医生在处理该类病人过程中,在出现肌无力的临床表现后,应积极进行对症处理,做好呼吸支持和营养支持,以降低或推迟致死性临床症状的发生。②有4例(30.8%)病人表现出文献中RRM2B基因突变所致的典型的肾小管疾病,但在晚发型病人未见到典型的肾小管病,该4例病人的腹部超声均表现为肾椎体高回声影,这可能与钙沉积相关。因此临床儿科医生应注意完善该类病人的肾脏超声,一旦发现肾脏有类似影像学表现,应考虑到RRM2B基因突变的可能性。③有6例(46.2%)病人的头颅影像学检查发现脱髓鞘样改变,除外7例未进行脑脊液检查的病人,6例行脑脊液检查的病人中脑脊液蛋白水平均明显升高,高度提示RRM2B基因突变可破坏血脑屏障;且编号为2、3的病人听力障碍均为应用抗生素药物后出现,猜测可能是因为血脑屏障受损导致药物穿透血脑屏障后作用于中枢神经系统,导致药物的中枢神经系统副作用发生率增加。因此,对于此类病人应谨慎使用中枢神经系统毒性的药物,以降低中枢神经系统副作用的发生的概率。④有7例(53.8%)病人出现不同的胃肠道症状(包括食欲差、呕吐、腹泻),其中编号为1、2、3、11的病人同时具备胃肠道症状、听力障碍、眼睑下垂/眼外肌麻痹,辅助检查头颅影像学检查异常、肌肉活检可见RRF等特征,临床上亦可诊断MNGIE,再次明确RRM2B基因突变可导致MNGIE的临床表型,扩充了MNGIE的基因谱。⑤在5例仍存活的病人中,包括2例临床表现严重的早发型病人,此2例病人在病程前期即给予G-管进行营养支持;2例呼吸肌辅助呼吸的病人在院外继续呼吸机辅助呼吸后仍可较长时间存活,提示对于此类病人,较早的进行营养学支持和必要的呼吸支持能显著改善病人预后。
在体外细胞实验中,转染pLVX-shRNA2-shRRM2B至293T细胞抑制细胞内RRM2B基因转录,转染Plvx-IRES-ZsGreen1-RRM2B(c.420G>C)模拟突变细胞,作为对照组转染pLVX-res-ZsGreen1-RRM2B挽救实验组的改变。实验结果证实,与对照组相比,含RRM2B基因c.420G>C的细胞RRM2B基因蛋白表达量显著下降,mtDNA含量下降,初步明确了该突变的致病性和致病机制。
在RRM2B基因变异的病人中:①肌无力的发生常意味着致死性临床症状的发生;②MNGIE的临床表型常见;③早期营养支持以及及时的呼吸支持可能会显著改善病人预后。
