
9个不同产地菊茎叶中多类型资源性化学成分的分析与评价△
2020-06-10 08:52常相伟魏丹丹宿树兰严辉赵明郭盛谢倩茹尚尔鑫钱大玮孙晓东段金廒
中国现代中药 2020年4期
常相伟,魏丹丹,宿树兰,严辉,赵明,郭盛,谢倩茹,尚尔鑫,钱大玮,孙晓东,段金廒*
1.中国药科大学 中药学院,江苏 南京 211198;2.南京中医药大学 江苏省中药资源产业化过程协同创新中心/ 国家中医药管理局中药资源循环利用重点研究室/中药资源产 业化与方剂创新药物国家地方联合工程研究中心,江苏 南京 210023;3.江苏鹤乡菊海现代农业产业园发展有限公司,江苏 盐城 224335
菊花为菊科植物菊ChrysanthemummorifoliumRamat.的干燥头状花序,是药食同源的大宗中药材,具有疏散风热、平肝明目、解毒消肿的功效[1]。随着我国菊资源产业链的拓展延伸,其采收过程中产生大量的根、茎、叶等废弃物,目前尚未得到充分利用[2],不仅造成资源的极大浪费,而且也增加了环境承载压力。因此,在当今社会资源愈来愈短缺的严峻形势下,开展菊非药用部位的综合利用和开发研究是亟待解决的科学及社会民生问题。研究表明,菊茎叶中同样含有种类多样、含量丰富的黄酮类、酚酸类、多糖类、挥发油类、核苷类、氨基酸类等多种资源性化学成分[3-5]。而且菊茎叶在历代本草中也有大量药用或食用的记载,如《本草纲目》称[6]:“(菊)其苗可蔬、叶可啜、花可饵、根实可药、囊之可枕、酿之可饮,自本至末,罔不有功”。《重修政和经史证类备用本草》[7]引《肘后方》治疔肿垂死方:“菊叶一握,捣绞汁一升,入口即活,此神验。冬用其根”。这些本草典籍的记载为菊茎叶的资源化利用提供了理论基础。
目前对菊花的研究多集中于其花序[8-10],而对其茎叶部位的研究则较少,菊非药用部位的资源价值尚未充分体现。亳菊、滁菊、贡菊、怀菊、杭菊、福白菊、祁菊、嘉菊等是我国菊花的主栽品种[11],基于资源化学的研究思路与方法,本研究系统地收集了产自安徽亳州、安徽滁州、安徽黄山、河南郑州、江苏射阳、浙江桐乡、湖北麻城、河北安国、山东嘉祥9个菊花产区的菊茎叶,采用现代分析方法对不同产地菊茎叶中总黄酮、总酚和可溶性糖类成分(总多糖、单糖和寡糖)等多类型资源性化学成分进行分析与评价,以期为菊茎叶资源的精细化利用与产业化开发提供数据支撑。
1 仪器与试药
1.1 仪器
Sartorius BT125D型电子分析天平(德国塞利多斯科学仪器有限公司);Enspire多功能酶标仪(美国Perkin Elmer 公司);KH-500DV 超声波清洗器(昆山禾创超声仪器有限公司);M67013涡旋振荡器(北京克格仪器有限公司);Anke LXJ-ⅡB离心机(上海安亭科学仪器厂);Milli-Q Integral 5超纯水系统(默克密理博公司);HH-S数显恒温水浴锅(金坛市医疗仪器厂);Waters 2695 Alliance高效液相色谱系统,配备自动进样器、四元泵溶剂系统以及EmpowerTM色谱工作站软件;Waters 2424型ELSD检测器。
1.2 试药
实验用水为自制超纯水;甲酸、甲醇、乙腈(色谱纯)(德国默克公司);对照品芦丁(中国食品药品检定研究院,批号:LGNR-5N77L);没食子酸(中国食品药品检定研究院,批号:G131992);标准品葡萄糖醛酸、葡萄糖、果糖、蔗糖、鼠李糖、甘露糖、麦芽糖、棉籽糖、水苏糖(阿拉丁试剂公司);其他化学试剂购自上海国药化学试剂公司,均为分析纯。
菊花茎叶样品于2018年10—11月采自安徽亳州、安徽滁州、安徽黄山、河南郑州、江苏射阳、浙江桐乡、湖北麻城、河北安国、山东嘉祥9个菊花产区,每个产地均采集10批次。经南京中医药大学段金廒教授鉴定为菊科菊属植物菊ChrysanthemummorifoliumRamat.的茎叶。不同产地菊茎叶标本存放于江苏省中药资源产业化过程协同创新中心。菊茎叶样品采集后,45 ℃烘干,粉碎后过80目筛,备用。不同产地菊茎叶样品信息见表1。
2 方法与结果
2.1 总黄酮和总酚含量测定
2.1.1对照品溶液的制备 取芦丁和没食子酸对照品适量,精密称定后分别置于10 mL量瓶中,加甲醇溶解后稀释至刻度,混匀。即得质量浓度分别为0.39、0.48 mg·mL-1的芦丁和没食子酸对照品储备溶液,4 ℃保存,备用。
2.1.2供试品溶液的制备 精密称定样品粉末1.0 g左右,按照1∶20(g·mL-1)料液比精密加入70%甲醇-水溶液,称质量,30 ℃超声提取1 h,补足减失质量。16 060×g离心15 min,取上清液,即得总黄酮和总酚含量测定供试品溶液,4 ℃保存,备用。
2.1.3标准曲线的建立
2.1.3.1芦丁标准曲线的绘制 采用硝酸铝-亚硝酸钠比色法[NaNO2-Al(NO3)3-NaOH]测定总黄酮含量,参考相关文献[3,12-14]对测定方法进行适当优化。精密吸取0、0.1、0.2、0.4、0.8、1.2、1.6、2.0 mL的芦丁对照品储备溶液,分别置于10 mL离心管中,各加纯水补足至2.0 mL,摇匀。再各加5%NaNO2溶液1.0 mL,摇匀,放置6 min。接着各加10%Al(NO3)3溶液1.0 mL,摇匀,放置6 min。再各加4% NaOH(1 mol·L-1)溶液5.0 mL,摇匀,放置15 min。以相应试剂作为空白对照,采用紫外-分光光度法在510 nm 处测定吸光度,以吸光度为纵坐标(Y),芦丁对照品溶液的质量浓度为横坐标(X)绘制标准曲线,得标准曲线回归方程Y=1.28X-5.50×10-3。结果显示芦丁在19.65~393.00 μg·mL-1吸光度与含量呈良好的线性关系(r=0.999 3)。
2.1.3.2没食子酸标准曲线的绘制 采用福林-酚法(Folin-Ciocalteu method)测定总酚含量,参考相关文献[13-15]对测定方法进行适当优化。精密吸取质量浓度为0.48 mg·mL-1的没食子酸对照品溶液0、10、40、60、80、100、150、200 μL,分别置于5 mL离心管中,各加纯水补足至200 μL。接着加1.5 mL Folin-Ciocalteu试剂,摇匀,放置4 min;随后再缓慢加入800 μL 7.5% Na2CO3溶液,摇匀,避光静置2 h。以相应试剂为空白对照,采用紫外-分光光度法在760 nm处测定吸光度,以吸光度为纵坐标(Y),没食子酸对照品溶液的质量浓度为横坐标(X)绘制标准曲线,得标准曲线回归方程Y=4.78X+1.45×10-2。结果显示没食子酸在23.80~476.00 μg·mL-1吸光度与含量呈良好的线性关系(r=0.999 4)。

表1 不同产地菊茎叶样品信息
2.1.4方法学考察
2.1.4.1精密度试验 按2.1.3.1和2.1.3.2项下方法,精密吸取1.0 mL芦丁对照品储备溶液以及0.2 mL没食子酸对照品储备溶液,分别依法连续测定6次吸光度。结果显示,总黄酮和总酚精密度试验的RSD分别为0.64%和0.59%,表明仪器的精密度良好。
2.1.4.2重复性试验 取射阳杭白菊茎叶样品粉末(SYHBJ-JY 1)6份,每份1.0 g左右,精密称定,按2.1.2项下方法制备总黄酮和总酚含量测定供试品溶液。再分别按2.1.3.1和2.1.3.2项下方法操作,测定吸光度,并分别计算其平均含量和RSD。结果显示,总黄酮和总酚平均质量分数分别为5.50、4.71 mg·g-1,RSD分别为1.91%和1.84%,表明方法的重复性良好。
2.1.4.3稳定性试验 精密吸取射阳杭白菊茎叶(SYHBJ-JY 1)供试品溶液2份,于0、4、8、12、24、48 h时分别按2.1.3.1和2.1.3.2项下方法操作,显色完成后立即测定吸光度。结果显示,总黄酮和总酚在48 h内RSD分别为1.75%和2.11%,表明供试品溶液在48 h内稳定。
另再取上述供试品溶液2份,分别按2.1.3.1和2.1.3.2项下方法操作,显色完成后分别在 0、15、30、45、60、90 min测定吸光度。结果显示,总黄酮和总酚在90 min内RSD分别为1.53%和1.89%,表明显色后的供试品溶液在90 min内稳定。
2.1.4.4加样回收率试验 取已知含量的射阳杭白菊茎叶样品粉末(SYHBJ-JY 1)6份,每份约0.5 g,精密称定后再分别精密加入芦丁对照品适量,按2.1.2项下方法制备总黄酮含量测定供试品溶液,并按2.1.3.1项下方法操作,测定吸光度,得总黄酮平均回收率为98.9%,RSD为1.83%,表明方法的准确性较好。另取相同样品粉末6份,分别精密加入没食子酸对照品适量,按2.1.2项下方法制备总酚含量测定供试品溶液,并按2.1.3.2项下方法操作,测定吸光度,得总酚平均回收率为100.7%,RSD为2.01%,表明方法的准确性较好。
2.1.5不同产地菊茎叶中总黄酮和总酚含量测定 精密吸取不同产地菊茎叶供试品溶液350 μL,各加纯水补足至2.0 mL,分别按2.1.3.1项下方法操作,测定吸光度,并根据标准曲线计算出不同产地菊茎叶中总黄酮的含量(以芦丁计)。另精密吸取不同产地菊茎叶供试品溶液50 μL,各加纯水补足至 200 μL,分别按2.1.3.2项下方法测定吸光度,根据标准曲线计算出不同产地菊茎叶中总酚的含量(以没食子酸计)。
不同产地菊茎叶总黄酮和总酚含量测定结果见表2。结果显示,不同产地菊茎叶中总黄酮含量存在较大差异,其中祁菊茎叶中总黄酮含量最高,高达(37.89±4.17) mg·g-1;而射阳杭白菊茎叶中总黄酮含量最低,仅为(5.52±0.94) mg·g-1。此外,不同产地菊茎叶中总酚含量同样存在较大差异,结果表明,祁菊茎叶中总酚含量最高,高达(17.12±2.00) mg·g-1;而滁菊茎叶和桐乡杭白菊茎叶中总酚含量相对较低,分别为(3.96±0.64)、(3.85±0.70) mg·g-1。

表2 不同产地菊茎叶中总黄酮、总酚和可溶性糖类成分含量测定结果 mg·g-1
注:—表示低于检测限而未检出。
2.2 总多糖含量测定
2.2.1对照品溶液制备 取葡萄糖、葡萄糖醛酸对照品适量,精密称定,分别置于10 mL量瓶中。加纯水溶解后稀释至刻度,混匀,即得到质量浓度分别为0.70、0.36 mg·mL-1的葡萄糖和葡萄糖醛酸对照品储备溶液,4 ℃保存,备用。
2.2.2供试品溶液的制备 取样品粉末约0.5 g,精密称定,置于100 mL具塞锥形瓶中。精密加入80%乙醇50 mL,90 ℃加热回流提取2 h,趁热滤过,弃去滤液,滤渣用热80%乙醇洗涤2~3次。滤渣连同滤纸置于锥形瓶中烘干,精密加入超纯水50 mL,100 ℃加热回流提取2 h[16-17]。16 060×g趁热离心10 min,取上清液,即得总多糖含量测定供试品溶液。
2.2.3标准曲线的建立 分别以葡萄糖和葡萄糖醛酸为对照品,参照文献[16-17]对中性和酸性多糖测定方法进行适当优化,采用苯酚-硫酸法测定不同产地菊茎叶中中性多糖含量,采用咔唑-硫酸法测定不同产地菊茎叶中酸性多糖含量。
2.2.3.1葡萄糖标准曲线的绘制 精密吸取0、0.1、0.2、0.4、0.6、0.8、1.0 mL葡萄糖对照品储备溶液,分别置于15 mL具塞试管中,加纯水补足至1.0 mL,混匀。另取纯水1.0 mL作为空白对照,平行操作。加入5%苯酚溶液2.0 mL,混匀,沿管壁缓缓加入浓硫酸7.0 mL,稍冷却后充分混匀,置沸水浴中加热20 min。然后迅速冷却至室温,混匀。采用紫-外分光光度法在490 nm处测定吸光度[16-17]。以吸光度为纵坐标(Y),葡萄糖的质量浓度(X)为横坐标绘制标准曲线,得回归方程Y=3.12X+4.40×10-3。结果显示,葡萄糖在70.1~701.0 μg·mL-1,吸光度与含量呈良好的线性关系(r=0.998 0)。
2.2.3.2葡萄糖醛酸标准曲线的绘制 精密吸取0、0.1、0.2、0.4、0.6、0.8、1.0 mL葡萄糖醛酸对照品储备溶液,分别置于10 mL具塞试管中,加纯水补足至1.0 mL,混匀。另取纯水1.0 mL作为空白对照,平行操作。加入12.5 mmol·L-1的四硼酸钠硫酸溶液5.0 mL,混匀,置沸水浴中加热10 min。趁热加入0.125%咔唑无水乙醇溶液0.2 mL,混匀,置沸水浴中加热15 min,迅速冷却至室温,混匀。采用紫外-分光光度法在512 nm处测定吸光度[16-17]。以吸光度为纵坐标(Y),葡萄糖醛酸的质量浓度(X)为横坐标绘制标准曲线,得回归方程Y=7.31X+3.46×10-2。结果显示,葡萄糖醛酸在35.5~355.0 μg·mL-1,吸光度与含量呈良好的线性关系(r=0.998 8)。
2.2.4方法学考察
2.2.4.1精密度试验 取2.2.1项下葡萄糖和葡萄糖醛酸对照品溶液各0.5 mL,分别按2.2.3.1和2.2.3.2项下方法操作,连续测定6次吸光度。结果显示,中性多糖和酸性多糖精密度试验的RSD分别为1.13%和1.24%,表明仪器的精密度良好。
2.2.4.2重复性试验 取射阳杭白菊茎叶(SYHBJ-JY 1)样品粉末6份,每份 0.5 g左右,精密称定,按2.2.2项下方法制备供试品溶液。分别按2.2.3.1和2.2.3.2项下方法操作,测定吸光度,并分别计算其平均含量和RSD。结果显示,中性多糖和酸性多糖的平均质量分数分别为49.30、20.19 mg·g-1,RSD分别为1.89%和1.99%,表明方法的重复性良好。
2.2.4.3稳定性试验 精密吸取射阳杭白菊茎叶(SYHBJ-JY 1)供试品溶液2份,于0、4、8、12、24、48 h时分别按2.2.3.1和2.2.3.2项下方法操作,显色完成后立即测定吸光度。结果显示,中性多糖和酸性多糖在48 h内RSD分别为1.94%和2.05%,表明供试品溶液在48 h内稳定。
另取上述供试品溶液2份,分别按2.2.3.1和2.2.3.2项下方法操作,显色完成后分别在0、15、30、45、60、90 min测定吸光度。结果显示,中性多糖和酸性多糖在90 min内RSD分别为1.75%和1.91%,表明显色后的供试品溶液在90 min内稳定。
2.2.4.4加样回收率试验 精密量取6份已知含量的射阳杭白菊茎叶供试品溶液各400 μL,分别置于15 mL具塞试管中,再分别精密加入0.70 mg·mL-1的葡萄糖对照品溶液200 μL,加纯水补足至1.0 mL,混匀,制成加样回收供试液。按2.2.3.1项下方法测定吸光度,得中性多糖平均回收率为103.0%,RSD为2.45%,表明方法的准确性较好。
另精密量取6份已知含量的射阳杭白菊茎叶供试品溶液400 μL置于10 mL具塞试管中,再分别精密加入0.36 mg·mL-1的葡萄糖醛酸对照品溶液200 μL,加纯水补足至1.0 mL,混匀,制成加样回收供试液。按2.2.3.2项下方法测定吸光度,得酸性多糖的平均回收率为104.1%,RSD为2.27%,表明方法的准确性较好。
2.2.5不同产地菊茎叶中总多糖含量测定 精密吸取不同产地菊茎叶供试品溶液800 μL,各加纯水补足至1 mL,分别按2.2.3.1和2.2.3.2项下方法操作,测定吸光度,并根据标准曲线计算出不同产地菊茎叶中葡萄糖及葡萄糖醛酸的含量。
总多糖含量=中性多糖含量+酸性多糖含量
(1)
测定结果见表2,结果表明不同产地菊茎叶中总多糖含量存在较大差异,其中怀白菊茎叶和射阳杭白菊茎叶中总多糖平均含量较高,分别高达(70.84±1.08) 、(70.32±2.90) mg·g-1;而嘉菊茎叶中平均总多糖含量最低,仅为(27.47±2.29) mg·g-1。
此外,中性多糖平均含量最高的为射阳杭白菊茎叶,高达(49.79±0.40) mg·g-1;中性多糖平均含量最低的为嘉菊茎叶,仅为(15.43±1.24) mg·g-1;酸性多糖平均含量最高的为怀白菊茎叶,高达(30.41±0.06) mg·g-1,而滁菊茎叶、桐乡杭白菊茎叶和嘉菊茎叶中酸性多糖含量相对较低。结果显示,祁菊茎叶中酸性多糖与中性多糖含量的比值为1∶0.86,其余产地菊茎叶中酸性多糖与中性多糖含量的比值在1∶1.19~1∶2.43,表明除祁菊外多数产地菊茎叶中中性多糖含量高于酸性多糖含量,中性多糖和酸性多糖均为菊茎叶中总多糖的重要组成部分,因此仅以单一的中性多糖或者酸性多糖含量作为指标评价菊茎叶中总多糖含量缺少全面性,需要将两者结合,综合评价。
2.3 单糖和寡糖含量测定
2.3.1对照品溶液制备 取8个单糖和寡糖对照品适量,精密称定,加水溶解制成质量浓度分别为鼠李糖(1,1.31 mg·mL-1)、果糖(2,1.28 mg·mL-1)、甘露糖(3,1.17 mg·mL-1)、葡萄糖(4,1.25 mg·mL-1)、蔗糖(5,1.23 mg·mL-1)、麦芽糖(6,1.33 mg·mL-1)、棉籽糖(7,1.19 mg·mL-1)、水苏糖(8,1.25 mg·mL-1)的混合对照品储备液,经0.22 μm的微孔滤膜滤过,4 ℃保存,备用。将上述制备的混合对照品溶液逐级稀释制成系列质量浓度,用于线性关系考察。
2.3.2供试品溶液制备 分别取不同产地菊茎叶粉末0.5 g,精密称定,置于50 mL 锥形瓶中。精密加入超纯水 20 mL,称质量,25 ℃超声提取 1 h,补足缺失质量。16 060×g离心15 min,取上清液,0.22 μm微孔滤膜滤过,取续滤液作为供试品溶液,4 ℃保存,备用。
2.3.3色谱条件 色谱柱:GRACE Prevail Carbohydrate ES 色谱柱(250 mm×4.6 mm,5 μm);流动相:水(A)-乙腈(B);梯度洗脱:(0~7 min,75%B;7~17 min,75%~55%B;17~19 min,55%~50%B;19~21 min,50%~75%B;21~25 min,75%B);流速1.0 mL·min-1;柱温25 ℃;进样量5 μL。ELSD漂移管温度80 ℃;增益8;载气流速2.5 L·min-1。
2.3.4方法学考察
2.3.4.1线性关系、最低检测限(LOD)和最低定量限(LOQ)试验 取混合对照品储备液,加纯水分别稀释制成系列质量浓度的混合对照品溶液,按2.3.3项下色谱条件进行HPLC分析。以峰面积的对数值为纵坐标Y,对照品溶液质量浓度(μg·mL-1)的对数值为横坐标X,进行线性回归分析,计算r。在信号对噪音比值(S/N)分别为3和10时测定LOD和LOQ。
线性关系、LOD和LOQ测定结果(见表3)显示:所测定的果糖、葡萄糖、蔗糖3个糖类成分在测定范围内均展现较好的线性关系(r>0.997 6),其LOD和LOQ值分别在18.30~19.00、38.40~40.00 μg·mL-1。
2.3.4.2精密度、重复性和稳定性试验 精密度试验:取混合对照品溶液,按2.3.3项下色谱条件,分别在1 d内重复进样6次和在连续3 d内重复进样3次。以测定各待测成分的峰面积,以各峰面积的RSD来评价日内及日间精密度。结果显示(见表4),日内精密度和日间精密度试验的RSD均小于3.2%,表明仪器的精密度良好。
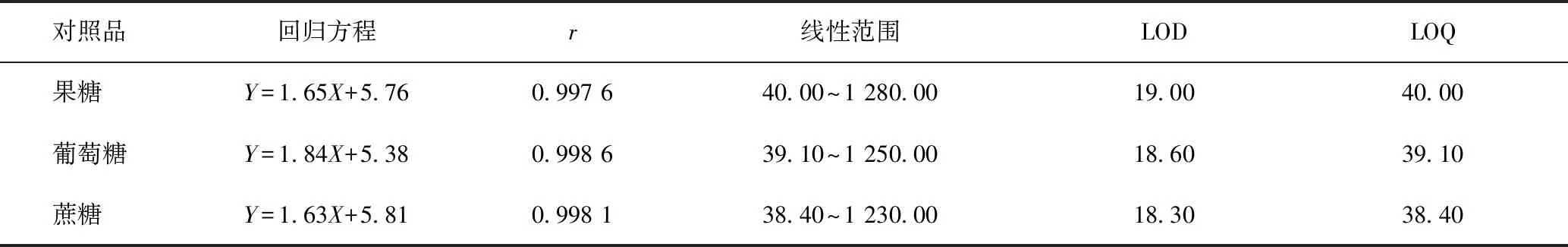
表3 果糖、葡萄糖和蔗糖线性关系、检测限和定量限 μg·mL-1
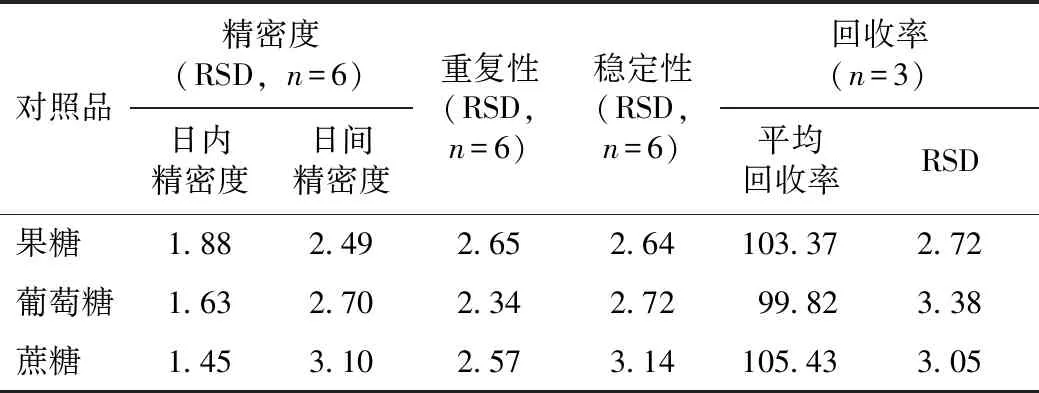
表4 精密度、重复性、稳定性和回收率试验结果 %
重复性试验:取福白菊茎叶样品粉末(FBJ-JY 1)6份,每份约0.5 g,精密称定,按2.3.2项下方法制备供试品溶液,经HPLC分析,计算供试品溶液中各待测成分的含量及其RSD。结果显示,果糖、葡萄糖、蔗糖的平均质量分数分别为94.04、33.18 和20.90 mg·g-1,RSD均小于2.70%(见表4),表明该方法重复性良好。
稳定性试验:取重复性试验中的一份供试品溶液,分别于0、4、8、12、24、48 h时注入液相色谱仪,计算各待测成分的峰面积及其RSD。结果显示(见表4),各待测成分峰面积的RSD均小于3.2%,表明供试品溶液在48 h内稳定性良好。
2.3.4.3加样回收率试验 取已知含量的福白菊茎叶样品粉末(FBJ-JY 1)9份,每份约0.25 g,精密称定,分别按样品中待测成分质量分数的50%、100%、150% 3个水平加入各对照品,每个水平平行测定3份,按2.3.2项下方法制备供试品溶液,并注入液相色谱仪按2.3.3项下色谱条件测定,计算平均回收率。结果显示(见表4),各待测成分的平均回收率为99.82%~105.43%,RSD均小于3.4%,表明该方法准确性较好。
2.3.5不同产地菊茎叶中单糖和寡糖含量测定 根据线性范围以及预实验结果,将上述制备的不同产地菊茎叶供试品溶液稀释2倍。应用所建立的分析方法分别测定9个不同产地菊茎叶样品中单糖和寡糖的含量。对照品及9个产地菊茎叶样品代表性HPLC-ELSD图见图1。结果显示(见表2),菊茎叶中含有的单糖类成分主要是果糖和葡萄糖,寡糖类成分主要是蔗糖(双糖)。其中,果糖是菊茎叶中主要的糖类成分,3种糖类成分平均含量由高到低依次为果糖、葡萄糖、蔗糖。相比果糖和葡萄糖,蔗糖在菊茎叶中的含量相对较低,仅在福白菊茎叶和祁菊茎叶中检测到,而在其他7个产地的茎叶中未能检测到。3种糖类成分在不同产地菊茎叶中的含量差异显著,其中尤以果糖为最,其平均质量分数最高可达(94.22±6.20) mg·g-1(福白菊茎叶),最低仅为(15.72±1.39) mg·g-1(嘉菊茎叶)。在9个产地的菊茎叶中,福白菊茎叶中果糖、葡萄糖和蔗糖的含量均最高,嘉菊茎叶中果糖和葡萄糖的含量均最低。湖北麻城产的福白菊茎叶呈高果糖含量特点,提示其可作为制备果糖的新资源,该研究结果为我国制糖战略资源的发现提供了理论基础。
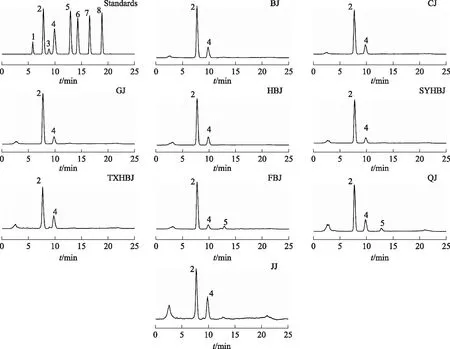
注:1.鼠李糖;2.果糖;3.甘露糖;4.葡萄糖;5.蔗糖;6.麦芽糖;7.棉籽糖;8.水苏糖。图1 对照品及不同产地菊茎叶样品溶液HPLC-ELSD图
2.4 菊茎叶中总黄酮、总酚、可溶性糖类成分以及种植区海拔之间的相关性分析
不同菊花种植区海拔变化较大,其中江苏省盐城市射阳县洋马镇海拔最低,仅为2 m;安徽省黄山市歙县北岸镇呈西村海拔最高,高达154 m。本研究利用R包“corrplot”对菊茎叶中总黄酮、总酚、可溶性糖类成分以及种植区海拔进行Pearson相关性分析,结果显示,海拔与菊茎叶中总黄酮、总酚、中性多糖、酸性多糖、总多糖、果糖和葡萄糖均呈正相关性,但是r<0.5,相关性较弱;总黄酮与总酚呈显著正相关性,r>0.9,表明菊茎叶中总黄酮和总酚积累趋势与规律相似;而中性多糖与总黄酮和总酚均呈负相关性。此外,可溶性糖类成分之间(中性多糖、酸性多糖、总多糖、果糖和葡萄糖)均呈较强正相关性,r>0.6,表明菊茎叶中可溶性糖类成分之间具有相互协同,促进积累的关系。
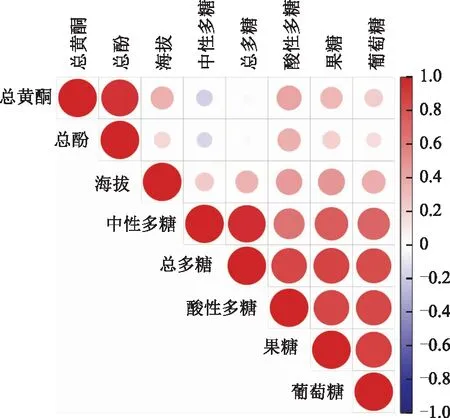
图2 菊茎叶中总黄酮、总酚、可溶性糖类成分 以及种植区海拔之间的相关性分析
3 讨论
中药资源类群中资源性化学成分的多途径、多层次、精细化利用是中药资源高效利用和可持续发展的重要策略[18]。本研究表明,菊茎叶中黄酮类、多酚类、可溶性糖类等资源性化学成分含量丰富,并且不同产地之间含量差异显著,据此可依据各类型资源性化学成分的潜在利用价值进行产业化开发,以提高菊茎叶的利用效率。黄酮和酚酸类成分是菊花中主要活性成分,具有抗菌[19]、抗炎[20]、抗病毒[21]、抗氧化[22]、心血管保护作用[23]等多种药理活性。本研究显示,祁菊茎叶中总黄酮和总酚含量均最高,分别为(37.89±4.17)、(17.12±2.00) mg·g-1,提示其具有开发成食品/化妆品天然抗氧化剂、饲料添加剂以及作为中兽药原料药的潜力。已有研究显示,菊花多糖具有多方面的生物活性和功能,尤其是对机体具有免疫调节功能[24]。本研究结果表明,怀白菊茎叶和射阳杭白菊茎叶中总多糖平均含量较高,分别高达(70.84±1.08)、(70.32±2.90) mg·g-1,提示其可作为功能性凉茶等保健产品开发的原料。研究结果尚显示,果糖是菊茎叶中主要的糖类成分。在甜度相同的条件下,能量值比葡萄糖、蔗糖的能量值低,且不易造成龋齿,是果糖广泛用于食品业中的主要原因,提示菊茎叶可开发作为一种功能性甜味剂,还能适用于糖尿病患者。此外,果糖在福白菊茎叶中含量最高,提示其可作为制备果糖的新资源。
综上所述,菊茎叶同样具有含量丰富的多类型资源性化学成分,本研究分析评价了我国不同产地菊茎叶中总黄酮、总酚以及可溶性糖类成分的含量差异,明确了不同产地菊茎叶中多类型资源性化学成分的组成与特点,为菊茎叶的资源价值发现以及合理综合利用提供了科学依据和参考。
猜你喜欢
电子测试(2022年16期)2022-10-17
食品工业(2022年1期)2022-02-21
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
汽车电器(2019年1期)2019-03-21
花火A(2018年9期)2018-11-26
江苏农业科学(2017年4期)2017-05-08
农业与技术(2016年24期)2017-04-20
