
福建省新型冠状病毒的分离和全基因组特征分析
2020-06-09 03:11:00张炎华何文祥吴冰珊王金章游丽斌黄枝妙陈宏彬俞婷婷鄢育青吴晶晶修文琼张小鸿翁育伟郑奎城
中国人兽共患病学报 2020年5期
张炎华,陈 炜,何文祥,吴冰珊,朱 颖,王金章,游丽斌,黄枝妙,陈宏彬,俞婷婷,鄢育青,吴晶晶,修文琼,袁 平,张小鸿,骆 婧,翁育伟,2,郑奎城,2
2019年12月以来,湖北省武汉市暴发了不明原因肺炎疫情,其他地区也陆续通报此类疫情。中国科学家在很短的时间内确定了该疾病系由一种新型冠状病毒所致。世界卫生组织将这种新型冠状病毒临时命名为2019-nCoV,并将2019-nCoV感染导致的疾病命名为COVID-19[1],我国卫生健康行政部门将该疾病命名为“新型冠状病毒肺炎”,简称“新冠肺炎”。2019-nCoV为β属的冠状病毒,该病毒有包膜,颗粒呈圆形或椭圆形,常为多形性,直径60~140 nm,基因组为单正链RNA,全长约30 kb,可编码E,M,N,S 4个主要的结构蛋白和若干个非结构蛋白。为了解福建省早期新冠肺炎病例中的2019-nCoV的基因特征,福建省疾控中心依托BSL-3实验室及时开展了病毒分离和高通量测序工作,并对病毒的全基因组序列进行了分析。
1 材料与方法
1.1 材 料
1.1.1样本来源 用于病毒分离的样本为福建省早期COVID-19确诊和疑似病例的呼吸道样本共12份,包括8份ORF1ab和N基因双靶标核酸检测阳性样本、1份N基因单靶标核酸检测阳性样本和3份双靶标核酸阴性样本。
1.1.2主要试剂与仪器 Vero-E6细胞(购自ATCC)、细胞培养基Minimum Essential Medium(Gibco,REF:41500-34)、胎牛血清(PANSera ES,cat:2602-P130707)、核酸提取试剂盒QIAamp Viral RNA Mini Kit(QIAGEN,cat:52906)、双链cDNA合成试剂Maxima H Minus Double-Stranded cDNA Synthesis Kit(ThermoFisher,cat:K2561)、文库构建试剂盒Ion Xpress Plus Fragment Library Kit(Thermo Fisher,cat:4471252)、模板制备与测序试剂盒Ion 520TM& Ion 530TMKit-Chef(Thermo Fisher,cat:A27757,与所选芯片型号配套)、测序芯片Ion 530TMChip Kit(Thermo Fisher,cat:A27763)、二氧化碳培养箱、倒置显微镜、Ion Torrent S5深度测序系统、序列拼接软件CLC Genomics Workbench (QIAGEN,ver.11.0)等。
1.2 方 法
1.2.1样本处理 COVID-19确诊和疑似病例的呼吸道样本中按照100∶1的比例加入10 000 IU的青霉素和10 000 μg的链霉素,使青霉素和链霉素的终浓度分别为100 IU和100 μg,置4 ℃过夜。
1.2.2细胞培养和病毒分离 将Vero-E6细胞接种至12.5 cm2的细胞瓶中,置36.0 ℃、5.0% CO2的培养箱中生长成单层细胞。吸去生长液(含10%胎牛血清),用病毒维持液(2%胎牛血清)清洗细胞2遍。每个细胞瓶加入0.5ml经双抗(青霉素和链霉素)处理的COVID-19患者样本,置36 ℃ CO2培养箱中吸附90 min后吸去样本并加入3.5 mL维持液,每日观察细胞病变情况。
1.2.3病毒核酸提取和反转录 核酸提取按照试剂盒说明书进行,提取的病毒核酸用于real-time RT-PCR检测;高通量测序的RNA在提取过程中加入QIAGEN公司RNase-Free DNase用于去除宿主细胞DNA。基因组反转录按照试剂盒Maxima H Minus Double-Stranded cDNA Synthesis Kit说明书进行。
1.2.4高通量测序 对分离阳性的样本进行高通量测序。样品经RNA提取、双链cDNA合成、双链cDNA纯化、双链cDNA定量、文库构建等步骤之后,采样Ion Chef System完成测序模板制备和530芯片上样,最后采用Ion Torrent S5测序仪进行高通量测序。利用CLC Genomics Workbench(ver. 11.0)软件拼接基因序列。
1.2.5序列分析 从GISAID和NCBI下载2019-nCoV基因组序列,利用MAFFT在线服务器做多序列排列(http://mafft.cbrc.jp/alignment/server/index.html),利用DNAStar MegAlign计算序列一致性,利用MEGA6.06软件构建基于全基因组序列以及S、E、M、N等结构蛋白编码序列的种系进化树(Neighbor Joining法,bootstrap=1000)。
2 结 果
2.1细胞分离培养结果 本次共接种12份咽拭子样本,包括8份ORF1ab和N基因双靶标核酸检测阳性样本、1份N基因单靶标核酸检测阳性样本和3份双靶标核酸阴性样本(见表1)。与正常细胞比较,8号和13号样本自接种后第3 d开始出现典型细胞病变(cyto-pathogenic effect,CPE),第6 d细胞完全病变。其他样本中,部分细胞形态发生改变,但与上述两份样本相比,CPE并不典型。为证实病毒分离成功,吸取培养物上清提取核酸做核酸检测,结果两份出现典型CPE的培养物Ct值较原始样本显著下降,3份接种前双靶标阴性样本核酸检测仍为阴性,其他样本有部分检出病毒核酸阳性,但Ct值大于样本接种前的Ct值,提示样本中的病毒未在细胞中增殖和复制,其核酸阳性应为样本残留所致,具体见表1。为进一步证实病毒分离结果,将所有样本第一代培养物继续盲传一代。结果显示除8号和13号样本出现典型CPE外(图1),其他样本均未出现CPE。

8号样本13号样本阴性对照(第2代第3 d,放大倍数:200倍)图1 Vero-E6细胞接种COVID-19患者咽拭子样本后的CPE情况Fig.1 CPE of Vero-E6 cell inoculated with throat swab specimens from COVID-19 patients
表1 新冠肺炎患者咽拭子样本分离培养及核酸检测情况
Tab.1 Nucleic acid test and CPE observation of Vero-E6 inoculated with throat swabs
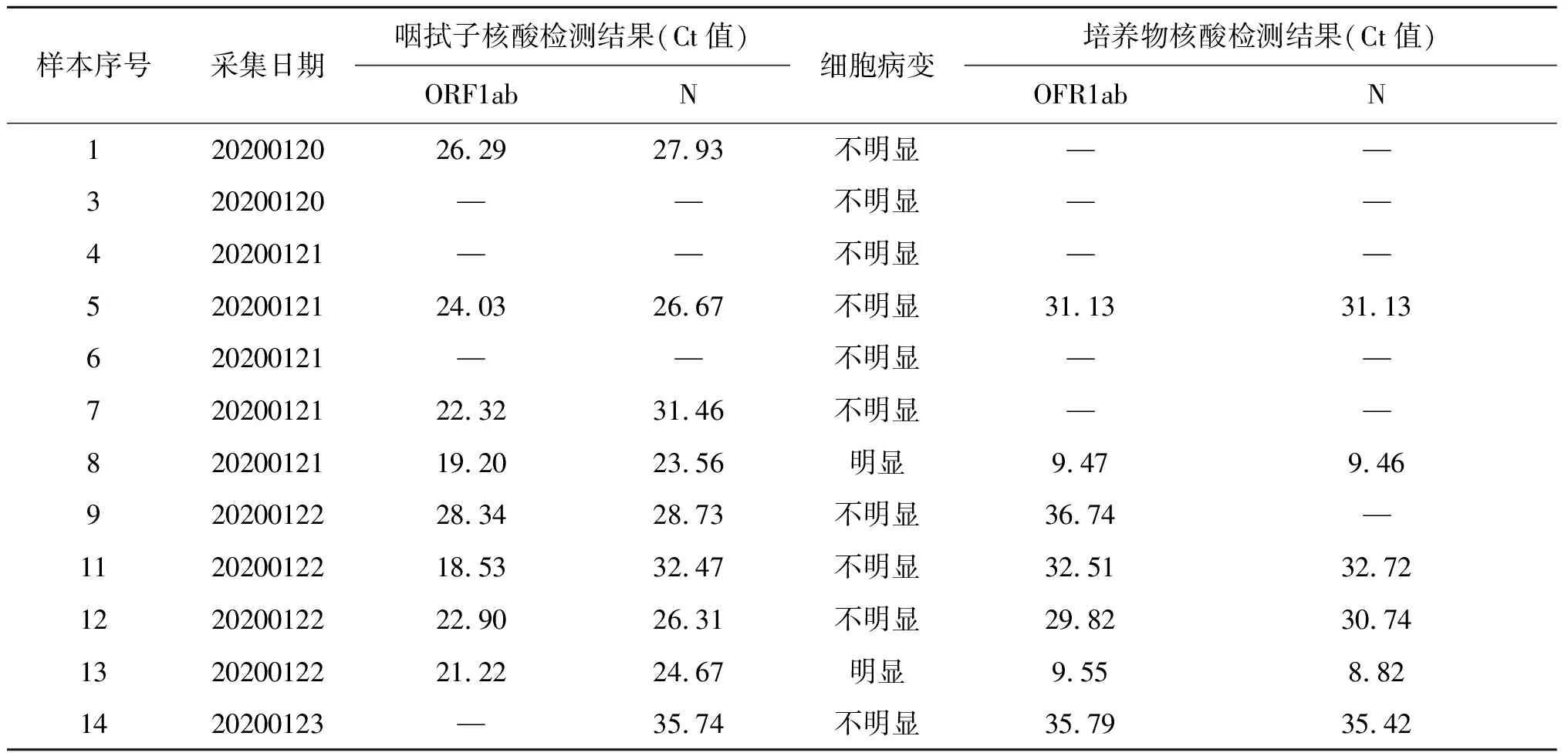
样本序号采集日期咽拭子核酸检测结果(Ct值)ORF1abN细胞病变培养物核酸检测结果(Ct值)OFR1abN12020012026.2927.93不明显——320200120——不明显——420200121——不明显——52020012124.0326.67不明显31.1331.13620200121——不明显——72020012122.3231.46不明显——82020012119.2023.56明显9.479.4692020012228.3428.73不明显36.74—112020012218.5332.47不明显32.5132.72122020012222.9026.31不明显29.8230.74132020012221.2224.67明显9.558.821420200123—35.74不明显35.7935.42
注:“—”代表阴性。核酸提取时均用50 μL RNase-free water洗脱。咽拭子样本的检测试剂为上海捷诺生物科技有限公司生产的新型冠状病毒检测试剂盒D2R(荧光定量PCR法),培养物的检测试剂根据中国疾控中心下发的检测方案[2],自行合成引物探针后配制反应体系。两种检测试剂的结果判定标准相同,Ct值<37为阳性。
2.2高通量测序结果 选取8号和13号样本培养物进行高通量测序。结果显示,两份样本所测定的总碱基数(Total Bases)分别为111 436 972和354 667 067,总reads数(Total Reads)分别为513 470和1 701 497,以早期公布的2019-nCoV病毒株Wuhan-Hu-1的基因组(GenBank Accession No.NC045512.2)为参考序列,所测reads的mapping数分别为445 476(86.76%)和1 113 346(65.43%),基因组平均测序深度(Average Coverage)分别为3169和7697。测序后的数据经CLC Genomics Workbench软件(ver.11.0)拼接,第8号样本培养物获得29 871 bp的基因组序列,命名为BetaCoV-Fujian-08-2020(GISAID No.EPI_ISL_411060),第13号样本培养物获得29 891 bp的基因序列,命名为BetaCoV-Fujian-13-2020(GISAID No.EPI_ISL_411066),两者基因组长度的差异主要在3′polyA的长度不同。
2.3基因组一致性分析 对BetaCoV-Fujian-08-2020和BetaCoV-Fujian-13-2020进行全基因组一致性分析,相对于Wuhan-Hu-1的一致性超过均99.9%,提示病毒尚未发生变异;相对于SARS-coronavirus(GenBank Accession No.NC004718.3)的一致性均为80.2%;相对于在蝙蝠身上分离到的病毒株BetaCoV_bat_Yunnan_RaTG13_2013 (GISAID No.EPI_ISL_402131)的一致性均为96.3%;相对于在穿山甲身上分离到的毒株BetaCoV_pangolin_Guandong_1_2019(GISAID No.EPI_ISL_410721)的一致性为90.4%和90.3%。从结构蛋白基因来看, 4个结构蛋白基因(E、M、N、S)中,两株2019-nCoV分离株的S基因与以上参考株的S基因的一致性最低。全基因组及E、M、N、S结构蛋白基因的一致性分析详见表2。
表2 福建省两株新型冠状病毒分离株与遗传相近的冠状病毒的核酸序列一致性分析
Tab.2 Nucleotide identity of two 2019-nCoV isolates from Fujian and genetic-related coronaviruses
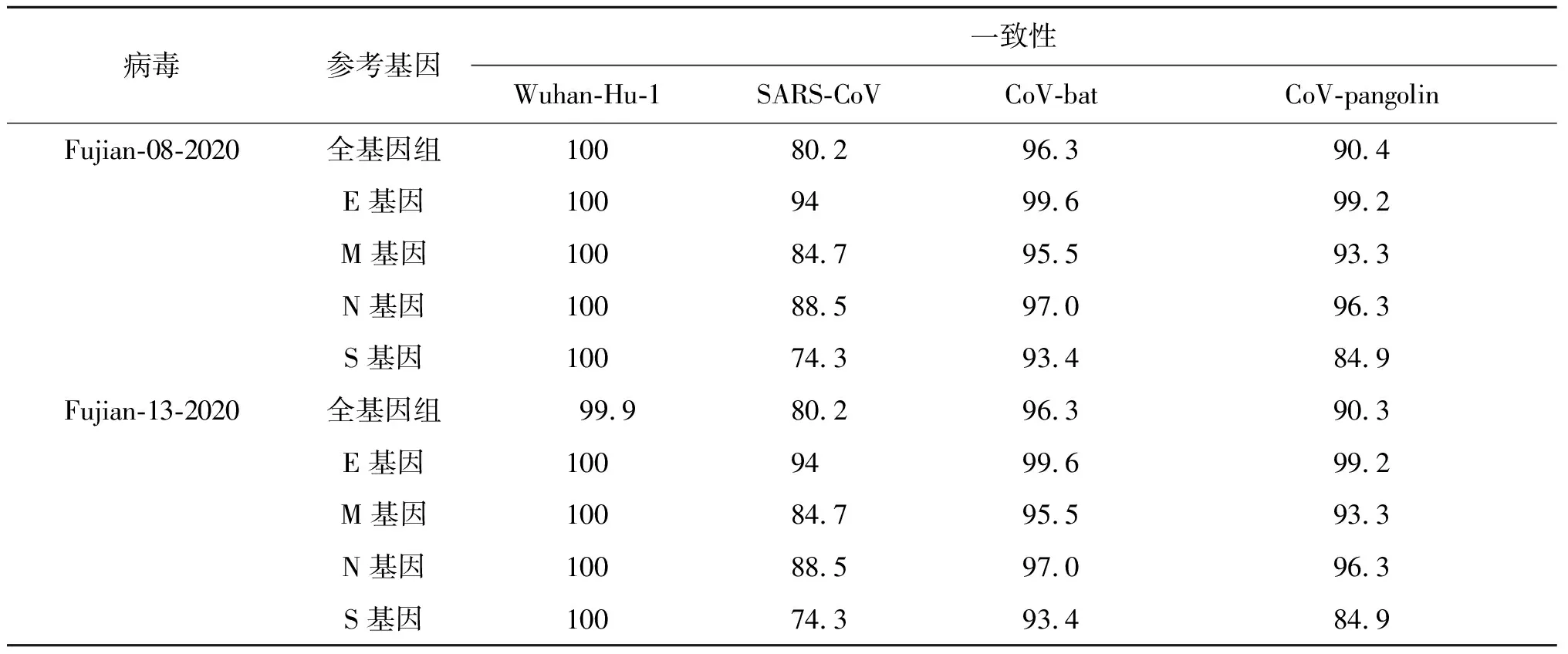
病毒参考基因一致性Wuhan-Hu-1SARS-CoV CoV-batCoV-pangolinFujian-08-2020全基因组10080.296.390.4E基因1009499.699.2M基因10084.795.593.3N基因10088.597.096.3S基因10074.393.484.9Fujian-13-2020全基因组99.980.296.390.3E基因1009499.699.2M基因10084.795.593.3N基因10088.597.096.3S基因10074.393.484.9
注:CoV-bat参比毒株为BetaCoV_bat_Yunnan_RaTG13_2013,CoV-pangolin参比毒株为BetaCoV_pangolin_Guandong_1_2019
2.4基因进化分析 全基因组进化分析显示,本次流行的2019-nCoV与2003年流行的SARS-CoV在同一个进化分枝上,亲缘关系比较近。研究结果显示,从福建省早期COVID-19病例中分离的BetaCoV-Fujian-08-2020和BetaCoV-Fujian-13-2020与国内其他省份以及其他国家发现的2019-nCoV在同一进化分枝上且亲缘关系非常相近。E、M、N、S结构蛋白基因的进化分析结果与全基因组进化分析结果基本相似,进一步提示病毒尚未发生明显变异。这些病毒与蝙蝠身上分离到的病毒BetaCoV_bat_Yunnan_RaTG13_2013的亲缘关系较穿山甲身上分离到的病毒BetaCoV_pangolin_Guandong_1_2019的关系更近。详见图2。
3 讨 论
国内已有学者报道SARS-CoV可在Vero、Ve S5高通量测序系统对培养物进行了基因组测序,将ro-E6、MDCK、Hela、Hep-2等不同细胞系中分离培养的情况[3],但是目前关于2019-nCoV在不同细胞系中分离培养敏感性的报道较少,为提高分离培养的效率,可以对不同细胞系对2019-nCoV的敏感性做进一步的研究。病毒在细胞中的分离培养情况与其结合到细胞受体上的能力有关。尽管2019-nCoV和SARS-CoV的受体均为血管紧张素转化酶(ACE2),但研究显示2019-nCoV的S蛋白中受体结合区域(Receptor binding domain, RBD)发生了氨基酸改变,提高了病毒与受体的亲和力[4-5],而且S1和S2连接区插入了PRRA 4个氨基酸,在连接区形成PRRAR序列[6],形成类似高致病性禽流感的HA链接肽,这或许会增强病毒的感染力。
通过对培养物进行核酸检测,我们初步确定分离到两株病毒。然而,两个基因靶标检测阳性尚不能完全确证分离到毒株,因此我们利用Ion Torrent测序结果进行拼接和比对之后获得了病毒的全基因组,证实了2019-nCoV分离培养获得成功。
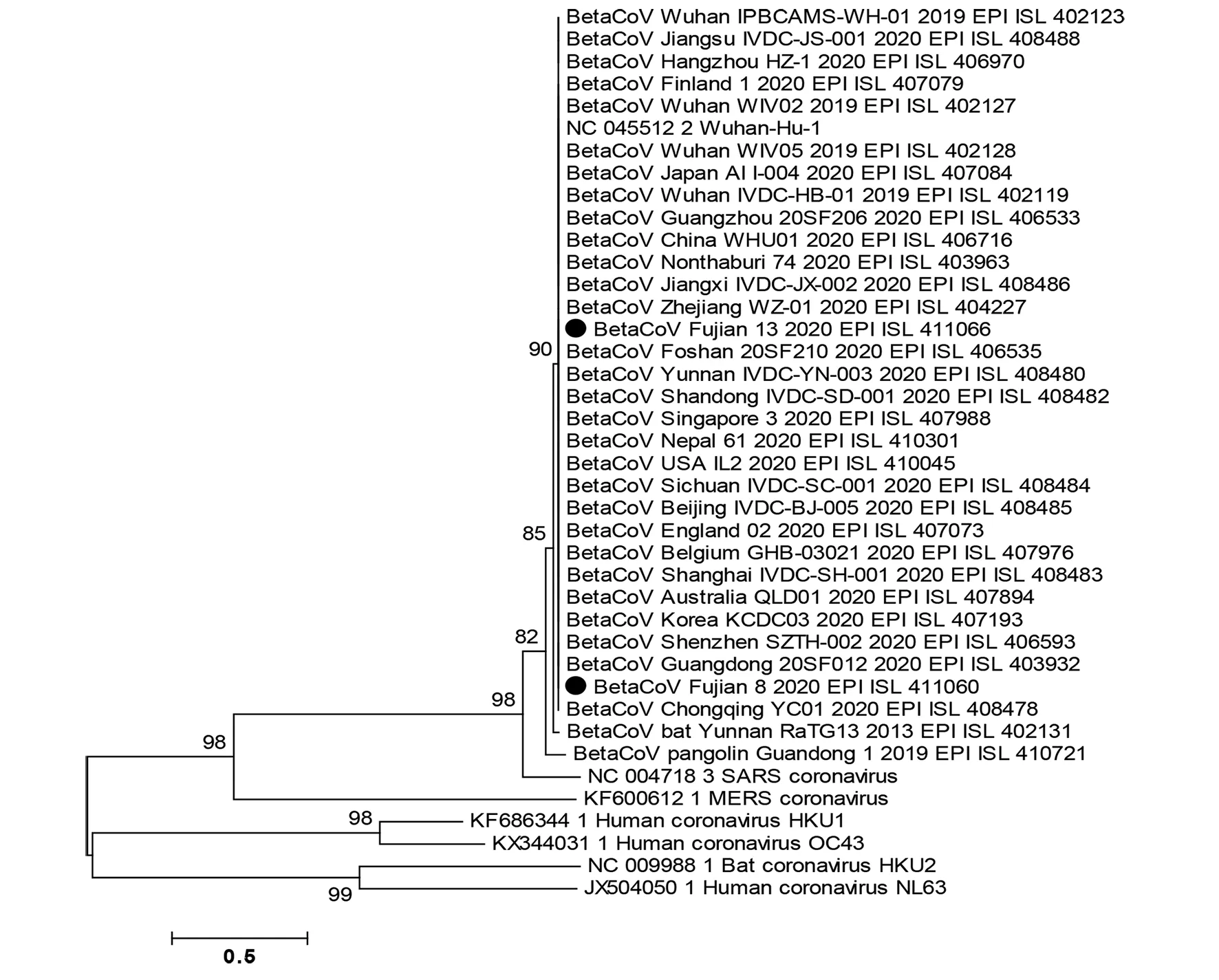
图2 福建省2株新型冠状病毒全基因组进化分析Fig.2 Phylogenetic tree of the complete genome of 2019-nCoV isolated from Fujian,China
通过对测序结果进行分析,我们发现自福建省早期COVID-19病例中分离到的2019-nCoV与Wuhan-Hu-1及其他地方的病毒全基因组一致性为99.9%以上,提示我省早期COVID-19患者所携带的病毒尚未发现变异。尽管本研究结果提示福建省分离到的2019-nCoV病毒没有发现明显的变异,但并不能排除病毒在疾病流行过程中逐步发生变异并导致生物学特征改变。在药物治疗、人体免疫和人际传播代次等因素的影响下,病毒变异很有可能随着时间延长而逐渐凸显。因此,我们应该对该病毒持续监测,密切关注病毒变异情况。
猜你喜欢
中华诗词(2022年9期)2022-07-29 08:33:50
公民与法治(2022年5期)2022-07-29 00:47:28
中国慈善家(2022年3期)2022-06-14 22:21:55
快乐语文(2021年34期)2022-01-18 06:04:14
教学考试(高考物理)(2021年5期)2021-11-08 10:31:22
今日农业(2021年11期)2021-08-13 08:53:24
中医眼耳鼻喉杂志(2021年1期)2021-07-22 07:38:14
中国(俄文)(2020年8期)2020-11-23 03:37:13
燕山大学学报(2015年4期)2015-12-25 02:19:49
遗传(2014年3期)2014-02-28 20:58:49
