
绿木霉(Trichoderma virens)T23甲基转移酶基因 gliN-T对胶毒素合成的调控研究
2020-06-04 01:37:48华丽霞蒋秋平曾华兰叶鹏盛王明娟杨晓丫何晓敏
中国农业大学学报 2020年6期
华丽霞 孙 佩 蒋秋平 曾华兰* 叶鹏盛 何 炼 曾 静 王明娟 张 敏 罗 飞 杨晓丫 何晓敏 刘 勇
(1.四川省农业科学院 经济作物育种栽培研究所,四川 成都 610300;2.农业农村部西南作物有害生物综合治理重点实验室,四川 成都 610066)
木霉菌(Trichodermaspp.)是研究和利用最广泛的植物病害生防真菌。已有研究表明木霉菌对黄瓜枯萎病病菌、辣椒疫霉病菌、棉花立枯丝核菌等多种植物病原真菌具有显著拮抗效果,抑菌率在80%以上[1-2];木霉菌能增强作物根系活力,对作物的生长具有促进作用[3-5]。因此,木霉菌在作物病害生物防治、农药减施增效方面具有重要的研究、开发及应用价值。胶霉毒素(Gliotoxin),简称胶毒素,是一种真菌次生代谢产物,因其对细菌、放线菌以及真菌等具有显著的拮抗作用,已被开发成抗生素或化疗药物[6]。木霉属中的部分菌种可产生胶毒素,该物质对病毒、细菌等均有抑制效果[7-8]。
胶毒素合成的分子机制在烟曲霉中研究较为深入:在烟曲霉基因组调控胶毒素合成的基因簇即gli基因簇中包含了12 个基因(gliA,gliC,gliF,gliG,gliK,gliM,gliN,gliJ,gliI,gliH,gliZ,gliT)共同调控着烟曲霉胶毒素的合成[9],目前,簇内gliP、gliC、gliZ、gliA、gliT在烟曲霉胶毒素合成途径中的作用已经较为清晰[10~14]。已有研究表明绿木霉菌(Trichodermavirens)中的部分菌株可产生胶毒素,并通过脉冲标记明确了绿木霉菌液体培养过程中胶毒素含量的动态变化情况[15],但是绿木霉菌胶毒素合成分子调控机制方面的研究远远滞后于烟曲霉胶毒素的研究。虽然Vargas等[16]对T.virens菌株Gv29-8中的gliP基因进行基因敲除,明确了该基因与绿木霉胶毒素的合成有关,然而,在分子水平上深入剖析绿木霉菌胶毒素合成的分子调控机制仍需要进行大量的科研工作。因此,开展绿木霉菌胶毒素合成基因的克隆及功能研究,有利于对胶毒素合成代谢的分子调控机制进行深入研究,有利于从分子水平上阐明木霉生防菌的拮抗机理,为进一步开发和应用提供重要的理论基础。
本研究室前期通过生物信息学方法,发现绿木霉菌T23基因组中存在1 个与烟曲霉胶毒素合成基因簇有同源性的基因簇,簇内有8 个候选基因,分别命名为gliP-T、gliC-T、gliN-T、gliK-T、gliI-T、gliG-T、gliF-T、gliM-T[17]。为进一步研究绿木霉菌胶毒素合成的分子调控机制,本研究拟以绿木霉菌T23为研究对象,通过改良的农杆菌介导的遗传转化(Agrobacteriumtumefaciens-mediated transformation, ATMT)技术,对T23胶毒素合成基因簇内编码带甲基转移酶结构蛋白的gliN-T基因进行敲除,分析基因敲除突变体中胶毒素含量变化情况,明确gliN-T对胶毒素合成的调控作用,为后续的胶毒素代谢调控机制的深入剖析提供重要的理论基础,同时为胶毒素的进一步开发利用提供参考信息。
1 材料与方法
1.1 供试材料
试验菌为生防绿木霉菌T23,由本研究室从川产道地中药材根际土样中分离得到[18]。
细胞壁裂解酶Glucanex(Sigma-Aldrich,L1412,美国);E.Z.N.A.®Fungal DNA Kit(Omega Biotek,D3390=01,美国);胶毒素标准品(1 mg,Lot number: 1D0E12, 纯度>99%,青岛普瑞邦生物工程有限公司);高效硅胶GF254预制板(青岛海洋化工有限公司);试验所需引物的合成及测序均由生工生物工程(上海)股份有限公司完成。
1.2 仪器设备及试剂
高效液相色谱1200型,配紫外检测器G1314B(安捷伦科技有限公司,美国);C18反相色谱柱(4.6 mm×250 mm, 5 μm, Pribolab, Singapore);色谱纯甲醇(美国Fisher);
1.3 培养基及相关试剂
马铃薯葡萄糖琼脂培养基(Potato dextrose agar, PDA)用于绿木霉菌T23的培养;LB培养基用于大肠杆菌及农杆菌的培养;IM液体诱导培养基用于ATMT诱导转化[19],121 ℃高温高压灭菌后,加入终浓度为200 μmol/L的乙酰丁香酮;共培养基用于农杆菌和分生孢子的共培养,其配方在IM培养基基础上,每升共培养基只需加入葡萄糖1 g,加入琼脂粉 15 g,121 ℃高温高压灭菌后加入终浓度为 200 μmol/L 的乙酰丁香酮,倒平板待用。
筛选培养基即PDA培养基高温高压灭菌后,加入终浓度为150 μg/mL的潮霉素B后倒平板待用。
1.4 基因敲除载体的构建及转化子的分子鉴定
1.4.1DNA 提取
将供试绿木霉菌株T23活化于PDA平板培养基上,培养2~3 d后,用接种环挑取新鲜菌丝接种到马铃薯葡萄糖液体培养基(Potato dextrose broth, PDB)中,28 ℃振荡培养2~3 d,待菌丝体生长茂盛的时候,用灭菌纱布和滤纸过滤收集菌丝体,用纸巾尽量吸干菌丝体中的水分后置于灭菌研钵中,加入液氮研磨至粉末状,然后用E.Z.N.A.®Fungal DNA Kit(Omega Biotek,D3390=01,美国)进行基因组DNA的提取,具体操作依照说明书进行。
1.4.2基因敲除载体构建及转化子的分子鉴定
按照同源敲除的原理构建基因敲除载体(图1):分别以gliN-T的5′端1 016 bp (LF)及3′端 892 bp(RF)作为同源臂;以构巢曲霉启动子PtrpC启动的潮霉素基因(hph),即潮霉素表达盒作为基因替换片段,按照LF::PtrpC-hph::RF的顺序,组装到携带有CaMV poly(A)终止子元件的基因敲除载体pGKO2中。根据PtrpC-hph序列设计正反方向引物,即hyp-F与hyp-R,用于转化子潮霉素盒的检测;同时根据LF序列设计正向引物gliN-F,根据RF序列设计反向引物gliN-R,用于同源重组转化子的分子鉴定。引物信息见表1。
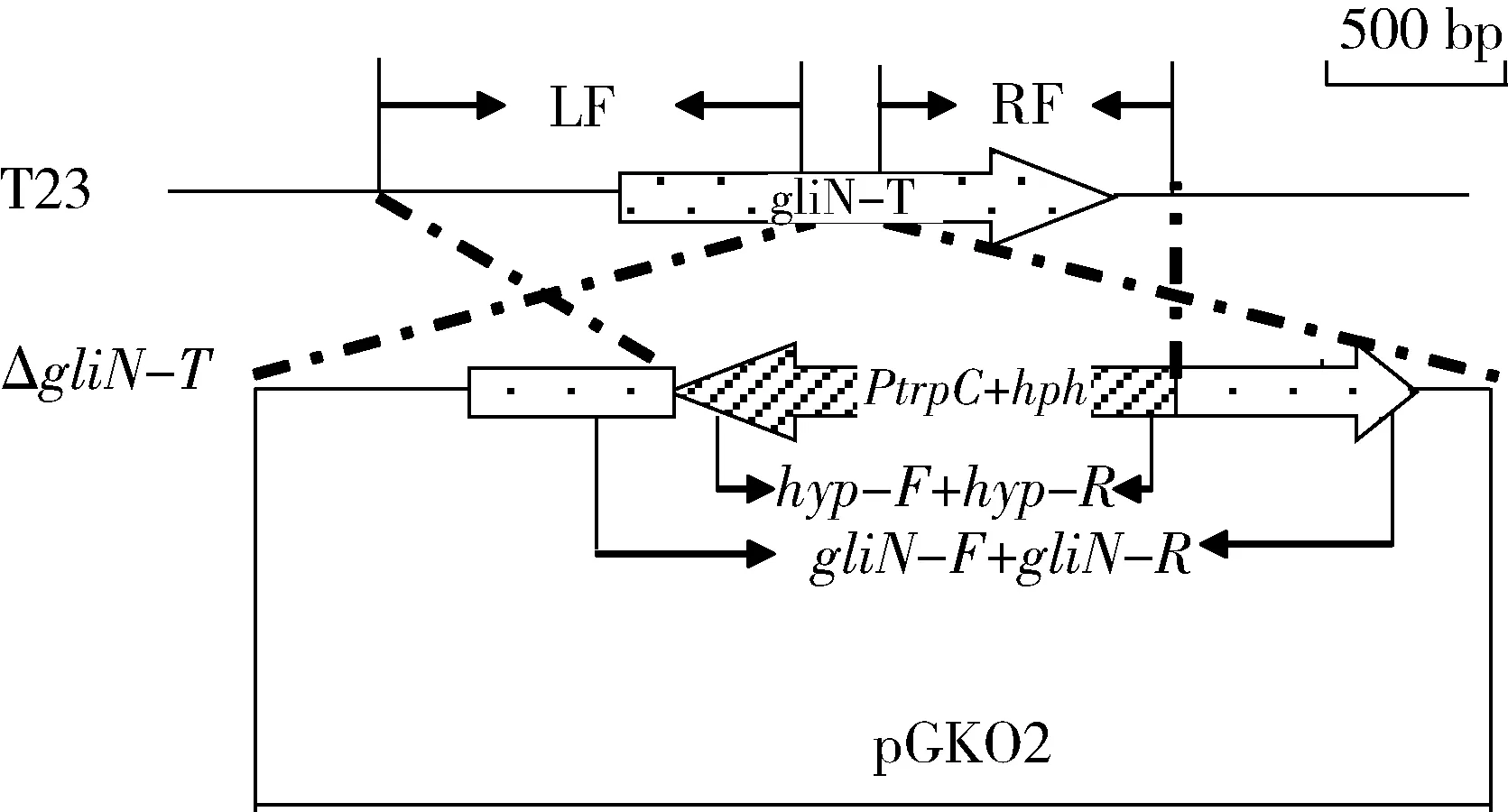
图1 基因敲除同源置换原理
Fig.1 Gene knockout strategy based on homologous recombination

表1 本研究所用引物Table 1 Primers used in this study
注:引物序列5′末端的小写字母为无缝拼接所需的同源重组序列;*,野生型和突变体扩增片段大小。
Note: Lowercase letters on the 5′-end present the homology of the adjacent fragment for vector construction using seamless cloning; *, length of PCR fragments of wild type and mutant.
1.5 农杆菌介导的遗传转化技术的改良
1)取携带有基因敲除构建体的阳性农杆菌C58C1在LB培养基上划线,28 ℃培养36 h,挑取单菌落到带抗生素的LB液体培养基中,28 ℃振荡培养过夜,离心收集菌体,随后用IM诱导培养基(含终浓度为200 μmol/L的乙酰丁香酮)重悬浮菌体并调节OD600至0.2~0.3,然后28 ℃振荡培养6 h,备用。
2)绿木霉分生孢子悬浮液的制备:在PDA培养基上活化绿木霉菌株T23,28 ℃培养4 d左右至产孢,用灭菌水轻轻地刮洗菌面,3 层擦镜纸过滤菌丝,4 000 r/m离心5~10 min收集分生孢子。倒掉上清,加入细胞壁裂解酶Glucanex(终浓度为15 mg/mL,溶于0.7 mol/L NaCl溶液),30 ℃ 80 r/m 振荡培养2.5~3.0 h,4 000 r/m离心,用0.7 mol/L NaCl溶液清洗1~2 遍,3 层擦镜纸过滤收集分生孢子液,调节分生孢子浓度至106个/mL。
3)取等体积(200 μL)的农杆菌菌液及孢子悬浮液进行混合后均匀涂布于共培养平板培养基中,22 ℃ 暗培养3 d。随后,在共培养好的平板上倒1 层含潮霉素(终浓度150 μg/mL)及头孢抗生素(终浓度400 μg/mL)的PDA筛选培养基,28 ℃培养 5 d 左右,挑取能在筛选培养基上生长的转化子到新的筛选培养基中,进行潮霉素稳定性鉴定。
1.6 基因表达分析
参照已有报道[17]提取野生型T23和基因敲除突变体ΔgliN-T的总RNA并进行反转录,合成cDNA第一链。在gliN-T基因外显子区域设计引物,在转录组水平上检测gliN-T在突变体中的表达情况。引物详细信息见表1。
1.7 胶毒素的提取及检测
1.7.1薄层色谱分析
将供试菌株接种到100 mL PDB培养基中,28 ℃ 震荡培养48 h,用孔径为0.22 μm的滤膜过滤培养液,取20 mL滤液,加入等体积的乙酸乙酯萃取2 次,合并萃取液,置于通风橱中挥干得到待测样品后用2 mL甲醇溶解作为薄层鉴别供试品。取胶毒素对照品1 mg,加入2 mL甲醇溶液,超声5 min,得到浓度为0.5 mg/mL的胶毒素标准品溶液,作为薄层分析的对照品溶液。以高效硅胶GF254预制板作为薄层板,以正己烷-乙酸乙酯体积比为6∶6作为展开剂,在254 nm的紫外光下观察薄层板的展开情况,随后用碘蒸汽对薄层板进行显色。
1.7.2高效液相色谱检测
挑取新鲜菌丝接种至200 mLPDB培养基中, 28 ℃中震荡培养48 h后取2 mL培养液并用0.22 μm 微孔滤膜过滤至进样瓶中待用。按照以下的色谱条件对培养液中的胶毒素进行检测:以水为流动相A,甲醇为流动相C,按照50%水加50%甲醇进行等度洗脱;流速:1 mL/min;柱温:30 ℃;进样量:20 μL;检测波长:254 nm。
2 结果与分析
2.1 通过改良的ATMT技术获得gliN-T基因敲除突变体
在细胞壁溶解酶Glucanex处理试验前,共进行了3 批次的遗传转化试验共筛选到3 个转化子。分子鉴定结果表明这3 个转化子均为异位整合转化子。在分生孢子液中添加终浓度为15 mg/mL的细胞壁溶解酶Glucanex进行处理后,筛选得到22 个转化子,潮霉素基因检测阳性率为100%,基因特异引物鉴定结果见图2:在22 个转化子中:有3 个是同源重组转化子,即成功敲除gliN-T的基因敲除突变体ΔgliN-T;有2 个转化子检测到潮霉素基因,但检测不到同源敲除的条带,为非正常转化子;其余17 个是异位整合转化子。对溶解酶处理前后的试验结果进行比较分析,发现绿木霉菌T23分生孢子经细胞壁溶解酶Glucanex处理后,平均每毫升分生孢子液的转化子数从0.25 个提升到10.00 个,转化效率大大提高(表2)。
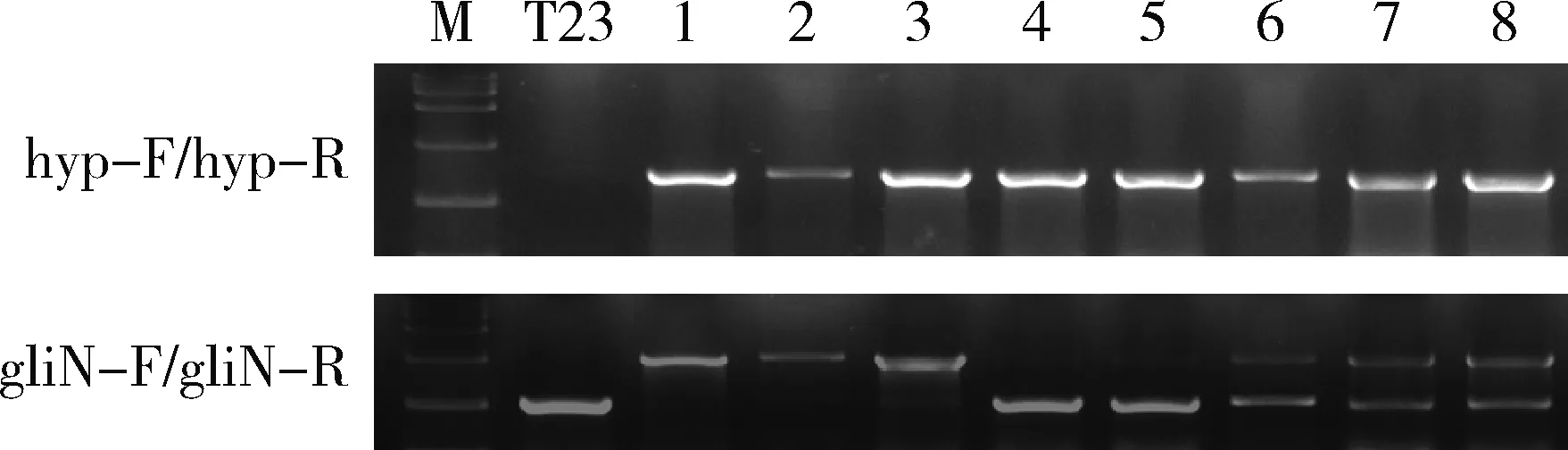
M:DL15000;T23:野生型T23 DNA扩增产物;1~8:基因敲除转化子DNA扩增产物(1~3为同源重组阳性转化子,4~5为非正常转化子,6~8为异位整合转化子)。 M: DL15000; T23: amplicon from wildtype T23 gDNA; 1-8 are amplicons from transformants gDNA: 1-3 represent homologous transformants; 4-5 are unusual transformants; 6-8 are ectopic transformants.
图2 Glucanex处理后获得的转化子的分子鉴定
Fig.2 Molecular identification for transformants after glucanex treatment
2.2 gliN-T在基因敲除突变体中的表达分析
对野生型T23及基因敲除突变体ΔgliN-T中的gliN-T进行RT-PCR表达分析,结果表明:在野生型T23基因组及cDNA中均检测到gliN-T的表达; 在转录水平上,在基因敲除突变体ΔgliN-T中检测不到gliN-T的表达(图3),意味着gliN-T基因被成功敲除。

表2 Glucanex处理前后木霉菌T23 ATMT转化效果比较Table 2 Comparison of transformant efficiency before and after Glucanex treated for T23
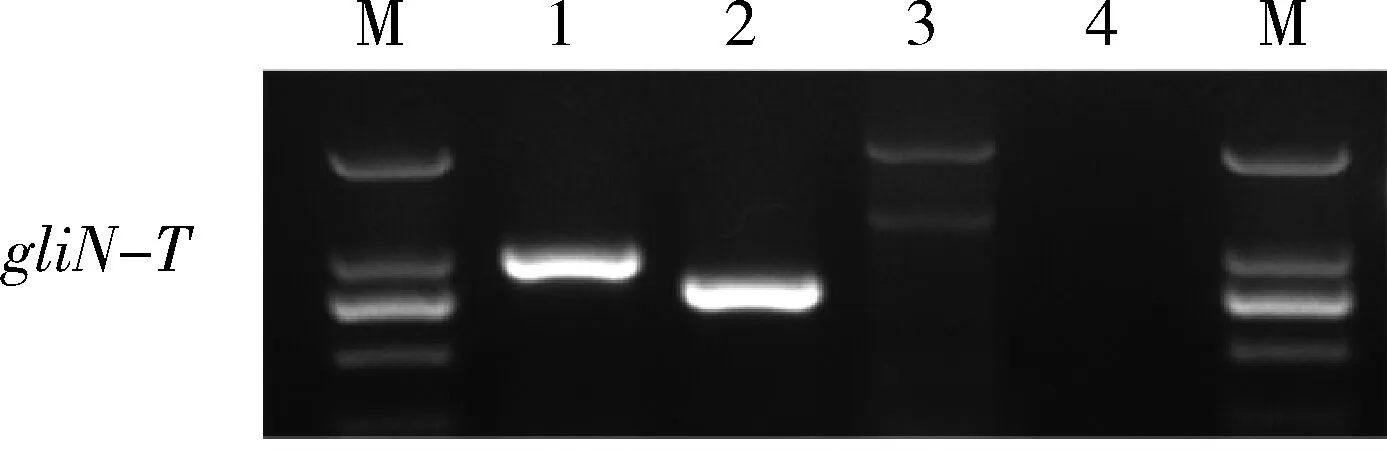
M:DL2000;1:T23 gDNA 扩增产物;2:T23 cDNA 扩增产品;3:ΔgliN-TcDNA 扩增产物;4:阴性对照,以H2O为模板的扩增产物。 M, DL2000; 1, amplicon from T23 gDNA; 2, amplicon from T23 cDNA; 3, amplicon fromΔgliN-TcDNA; 4, amplicon from H2O as negative control.
图3gliN-T在基因敲除突变体ΔgliN-T中的表达分析
Fig.3 Expression analysis forgliN-Tin gene knockout mutantΔgliN-T
2.3 gliN-T基因敲除突变体胶毒素代谢产物的检测
对野生型与基因敲除突变体ΔgliN-T的48 h的培养液进行薄层色谱分析,胶毒素标准品上样量为5 μL,供试样品上样量为10 μL。薄层色谱分析结果表明,在野生型T23 48 h培养液中可检测到胶毒素(图4,泳道2和4);而相同培养条件下,基因敲除突变体ΔgliN-T的培养液则无法检测到胶毒素(图4,泳道3)。
为了进一步明确胶毒素的分泌是否与菌丝生长时期有关,本研究对不同培养时期(24、48、72和96 h)的野生型T23及基因敲除突变体ΔgliN-T的菌丝培养液分别进行了高效液相色谱检测。检测结果发现,在28 ℃,120 r/m的培养条件下,培养24 h时,野生型T23培养液中检测不到胶毒素,在48、72、96 h的培养液中均检测到胶毒素;而在相同的培养条件下,基因敲除突变体ΔgliN-T在4 个生长时期的培养液中均未检测到胶毒素(表3)。

(a) 薄层板在254 nm紫外光下的薄层检测结果;(b) 经过蒸汽碘显色后的薄层色谱图 1和5:胶毒素标准品;2和4:野生型T23培养液萃取样品;3:突变菌株培养液萃取物。 (a) chromatography under 254 nm UV light; (b) chromatography after vapor iodine staining 1 and 5, gliotoxin standard substrate; 2 and 4, extraction from wild type T23; 3, extraction fromΔgliN-T.
图4 薄层色谱法检测T23及突变体胶毒素
Fig.4 Gliotoxin detection by TLC for T23 andΔgliN-T
表3 高效液相色谱检测不同时间段培养液中的胶毒素含量
Table 3 Gliotoxin detection for different time-period culture filtrate by HPLC mAU·S
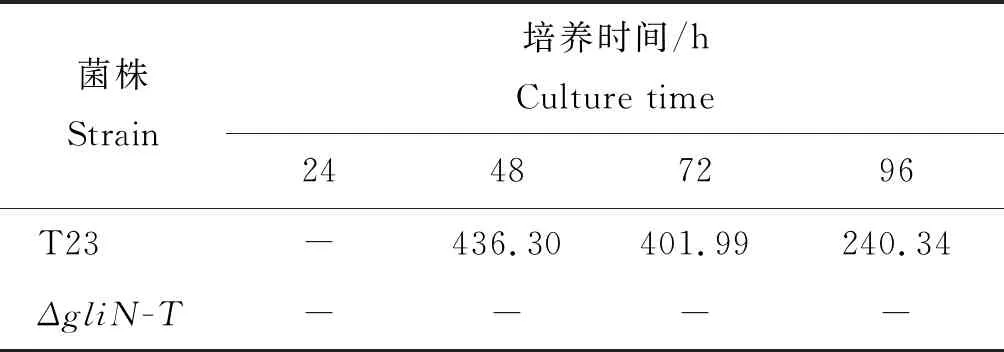
菌株Strain培养时间/hCulture time24487296T23-436.30 401.99240.34ΔgliN-T----
注:“-”表示在相同液相色谱条件下未检测到峰值。
Note: - represents no peak is detected under the same HLPC condition.
3 讨论与结论
高效率的基因敲除技术是进行丝状真菌基因功能研究的重要生物技术之一。目前应用于丝状真菌基因敲除的转化技术包括分生孢子电击转化[20]、基于菌丝原生质体的PEG/CaCl2转化[21]、基因枪转化及农杆菌介导的遗传转化(ATMT)[22]等。其中,ATMT技术因成本低、操作简单、转化效率高、遗传稳定等优点已经被广泛应用到各类丝状真菌的遗传转化中[23]。农杆菌介导的遗传转化技术虽然已经在绿木霉菌(T.virens)中有成功应用的报道[24-25],但是转化效率低,大大限制了T.virens重要基因的功能验证及开发利用。本研究对ATMT技术进行改良,在T.virensT23的分生孢子液中添加细胞壁溶解酶,转化效率是对照组的40 倍,并通过改良的ATMT技术获得胶毒素合成候选基因gliN-T的基因敲除突变体,这将为深入研究T.virensT23胶毒素合成的分子调控机制提供了强有力的技术支撑。
甲基转移酶是一类普遍存在于生物体内的重要酶类,参与生物体的甲基化反应,对生物体次生代谢产物的合成具有重要的调控作用[26-27]。在烟曲霉胶毒素合成基因簇内存在着2 个甲基转移酶基因(gliN及gliM),但是,尚未见有关烟曲霉胶毒素合成基因簇内甲基转移酶基因的功能研究报道。
本研究利用改良的ATMT技术对gliN-T进行基因敲除并获得突变体,发现敲除gliN-T基因后,T.virensT23胶毒素的合成受到抑制,gliN-T参与了调控T23胶毒素的合成。在胶毒素合成基因簇内存在着gliN-T及gliM-T2 个甲基转移酶基因,这2 个基因甲基化底物是否相同?其在调控胶毒素合成途径中各扮演着什么角色?这些问题有待深入探索。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:30
中国森林病虫(2021年6期)2021-12-20 08:45:42
中国森林病虫(2018年4期)2018-09-19 12:13:08
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
少儿科学周刊·少年版(2015年3期)2015-07-07 21:12:55
少儿科学周刊·少年版(2015年3期)2015-07-07 21:11:11
塔里木大学学报(2015年1期)2015-04-25 02:38:44
山东医药(2015年40期)2015-02-28 14:28:45
大豆科技(2014年5期)2014-03-23 02:46:18
