
色素血管性斑痣性错构瘤病4例临床分析及文献复习
2020-06-01 10:09梁文蒋雨薇方蔷
中国美容医学 2020年3期
关键词:临床表现
梁文 蒋雨薇 方蔷

[摘要]探讨色素性血管斑痣性错构瘤病患者的临床分型、发病机理、伴随系统症状以及治疗。通过报道笔者科室收集的4例色素血管性斑痣性错构瘤病,进行临床症状分型、常规检查、激光治疗并复习相关文献。发现4例患者中1例属于Cesioflanme型,1例对应Ⅱa型,1例对应Ⅱb型,2例归于Spilorosea型对应Ⅲa。GNA11和GNAQ基因突变可能导致了疾病发生。常见合并症有Sturge-Weber综合征、Klippel-Trenaunay综合征、眼球黑变病等。激光治疗后皮损外观改善。因此,色素血管性斑痣性错构瘤病因其临床表现及合并系统症状的多样性极易误诊漏诊。一旦发现,应进行系统检查。早诊断行激光治疗能够提高患者生活质量。
[关键词]色素血管性斑痣性错构瘤;临床表现;临床分型;发病机理
[中图分类号]R732.2 [文献标志码]B [文章编号]1008-6455(2020)03-0059-04
Abstract: The clinical classification,pathogenesis, concomitant systemic symptoms and treatment of patients with Phakomatosis Pigmentovascularis were investigated. Four cases of Phakomatosis Pigmentovascularis were collected by our department. The clinical symptoms were classified. Routine examination and laser treatment were completed . Related literature were reviewed. Four cases of patients with type 1 and 3 belong to Cesioflanme. Type 1 correspond Ⅱa. Type 3 correspond Ⅱb. Cases of type 2 and 4 belong to Spilorosea corresponding Ⅲ a. Mutations in the GNA11 and GNAQ genes may have caused the disease. Common complications include Sturge-Weber syndrome, Klippel-Trenaunay syndrome and eye melanosis. The appearance of skin lesions was improved after laser treatment. The variety of clinical manifestations and combined systemic symptoms of Phakomatosis Pigmentovascularis is easily misdiagnosed and missed diagnosis. The system should be checked. Early diagnosis and laser therapy can improve patients' quality of life.
Key words: Phakomatosis Pigmentovascularis; clinical manifestations; clinical classification; pathogenesis
色素血管性斑痣性錯构瘤病 (Phakomatosis Pigmentovascularis,PPV) 是一种先天性毛细血管畸形与色素沉着病变(如:色素性毛表皮痣、斑痣、咖啡斑和真皮黑色素细胞增多症) 为特征的罕见综合征[1]。患者可以仅表现为皮肤状况也可表现皮肤外症状。由于临床上对此病认识不足,容易漏诊误诊。本研究回顾性分析笔者科室2017年7月-2019年7月收集的4例患者临床资料并进行总结分析。
1 临床资料
1.1 一般资料:病例1:男,26岁。因面部大片红色和青褐色斑片20年至笔者科室就诊。患者出生时发现右侧面颊部及巩膜褐青色斑,额头鼻梁红斑,皮疹随年龄增长逐渐扩大,无自觉症状。既往体健,智力正常,家族中无类似病史。皮肤科检查:额头、面颊部、口周大面积褐青色斑片,鼻梁,面颊近鼻翼处暗红色斑片、斑块。双侧巩膜黑色斑片。实验室检查:血、尿及粪常规无异常,肝肾功能无异常,胸部正位X片无异常,肝、胆、胰、脾B超无异常,常规心电图无异常。治疗:面部褐青色斑片采用Nd:YAG 1 064nm激光治疗,间隔3个月1次,共行8次治疗,皮损明显消退。红色斑片采用595nm脉冲染料激光治疗,每2个月1次,共行8次治疗,红色有所减淡,仍有红斑(见图1)。
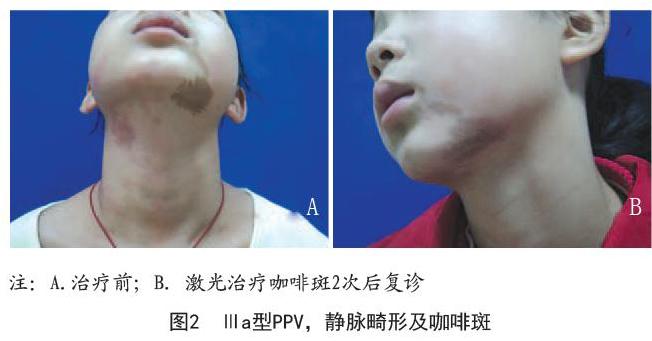
病例2:女,10岁。因面部红色和褐色斑片10年至笔者科室就诊。患者出生时发现右侧面颊部颈部淡红色斑片,左侧口角可见褐色斑片。既往体健,家族中无类似病史。皮肤科检查:右侧面颊部颈部手掌大小淡红色斑片,左侧口角可见褐色斑片,边缘形态不规则,界线清楚。实验室检查:血、尿及粪常规无异常,肝、肾功能无异常,胸部正位X片无异常,肝、胆、胰、脾B超无异常,常规心电图无异常。口角咖啡斑采用Nd:YAG 532nm激光治疗,间隔2个月1次,共行3次治疗,皮损有所减淡。鲜红斑痣采用595nm脉冲染料激光治疗,2个月1次,共行3次治疗,红色消退明显。见图2。
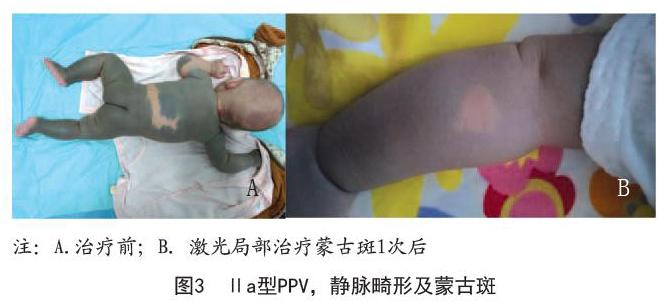
病例3:女,3月22天。因全身红黑色皮疹3月余就诊。患者出生时发现头皮、躯干、四肢弥漫性黑青色斑片,头皮、项部红色斑块。1个月前头皮出现新发黑青色斑片,头皮红斑颜色有所减淡,无自觉症状。饮食、睡眠、大小便尚可。家族中无类似病史。皮肤科检查:头皮、躯干、四肢弥漫性黑青色斑片,后腰处可见带状正常皮肤,头皮沿正中线可见鲜红色斑片,项部正中可见直径1.5cm鲜红色斑块,稍凸起皮面,表面结痂。实验室检查:血、尿及粪常规无异常,肝肾功能无异常,胸部正位X片无异常,肝胆胰脾B超无异常,常规心电图无异常。右前臂褐青色斑片采用Nd:YAG1 064nm调Q激光治疗小面积观察,间隔3个月1次,已治疗1次,色斑颜色明显减淡。头皮鲜红斑痣待观察,如无明显消退,后期可行595nm脉冲染料激光治疗。见图3。
病例4:男,18岁。因右上肢鲜红色斑片18年就诊。患者出生1月时发现右上肢有鲜红斑片,随年龄逐渐增大。10年前面部躯干出现散在淡褐色斑片。既往体健,智力正常,性格内向。皮肤科检查:右侧上肢大面积鲜红色斑片,边缘不规则,界线清楚。面部散在粟粒大小淡褐色斑点,前胸后背泛发直径0.2~2.0cm淡褐色类圆形斑片。实验室检查:血、尿及粪常规无异常,肝肾功能无异常,胸部正位X片无异常,肝胆胰脾B超无异常,常规心电图无异常。右上肢鲜红斑痣采用595nm脉冲染料激光治疗5次,2个月1次,红色消退明显。躯干咖啡斑面积广泛,未治疗,需观察有无皮肤神经纤维瘤改变。见图4。
1.2 色素与血管性病变的镶嵌方式:血管性和色素性病变在皮肤上的镶嵌方式中,2例呈斑块样相互融合,没有中线分界现象。其中血管性疾病占优势者2例,色素性疾病占优势者1例,两者均等者1例。
1.3 PPV伴发的疾病:病例1有眼黑变病,其余病例暂没有发现皮肤以外的异常。
2 讨论
2.1 临床分型:PPV的血管性改变主要是静脉畸形,表现为鲜红斑痣。色素性异常可表现为色素性毛表皮痣,不同于色素痣分型里的表皮痣。色素细胞活跃在表皮层的咖啡斑、斑痣。色素异常增生在真皮层的太田痣、伊藤痣、蒙古斑。根据色素性皮损与血管畸形的关系,可以分为不同类型的PPV。
传统临床分型将PPV分为四型,I型为微静脉畸形伴色素性毛表皮痣,又称Becker痣,临床少见,Joshi等[2]在1999年报道了1例16岁男孩,同时具有鲜红斑痣、Becker痣、咖啡斑,认为符合I型PPV诊断。黄玉成等[3]报道15例PPV患者,其中1例患者为鲜红斑痣并发Becker痣及白癜风,归为I型。Ⅱ型为微静脉畸形伴真皮色素斑,如:太田痣、伊藤痣或蒙古斑,伴有或不伴有贫血痣。Ⅲ型为微静脉畸形伴斑痣,也可有贫血痣。Ⅳ型为微静脉畸形伴斑痣及真皮色素斑,伴有或不伴有贫血痣。第V型为先天性毛细血管扩张性大理石样皮肤(cutis marmorata talangiectatic congenita)合并真皮色素斑。根据有无皮肤外系统损害,色素血管性斑痣性错构瘤又分为a亞型无系统损害,b亚型有皮肤外系统损害。因此色素血管性斑痣性错构瘤病按照临床传统分为5型,10个亚型[3]。
2005年,Happle[4]提出新的分类方法认为色素性毛表皮痣不是起源于色素细胞,故应排除I型,Ⅳ型又非常罕见,故建议把PPV分为四型。Cesioflanme型为微静脉畸形伴真皮色素斑(太田痣、伊藤痣和蒙古斑),对应于传统分类的Ⅱa和Ⅱb型;Spilorosea型为微静脉畸形伴斑痣,对应于传统分类的Ⅲa和Ⅲb型;Cesiomarmorata型为先天性毛细血管扩张性大理石皮肤伴真皮色素斑,对应于传统分类的Va和Vb型。最后一型为未能分类的PPV型。
本文中病例1、病例3为鲜红斑痣分别合并太田痣和蒙古斑,因此属于Cesioflanme型。病例1合并巩膜黑斑,对应Ⅱb型;病例3对应Ⅱa型。病例2、病例4无系统症状,为鲜红斑痣合并咖啡斑,归于Spilorosea型对应Ⅲa。
2.2 发病机理:色素血管性斑痣性错构瘤病是由于胚胎期神经嵴血管舒缩神经细胞和黑素细胞发育异常所致[5]。非等位基因双生斑是指在正常细胞的背景下,通过体细胞重组,由胚胎相邻细胞的两个基因不同克隆组成的双生斑点。Happle[6-7]曾认为在遗传上PPV是由于双生斑(twin-spotting)的现象导致了血管和色素的同时异常,是由于杂合性的丧失而引起的。
Nanda等[8]认为皮肤病变的散在性和镶嵌分布提示后合子突变。色素性角化性斑痣性错构瘤(phacomatosis pigmentokeratotica ,PPK)是早期提出的非等位基因双斑的一个例子。在PPK中,Groesser等[9]发现合子后激活的HRAS(哈维大鼠致癌基因同源物)在多能祖细胞中发生突变,可同时引起皮脂腺痣和黑素细胞痣,从而证明这是一种镶嵌性皮肤病。这一观察结果否定了在这种情况下非等位基因双斑点的假设,认为PPK最好通过多能祖细胞中的后合子激活HRAS突变来解释。随着研究的进展,Happle现在也提出色素血管性斑痣性错构瘤病是一种伪双生斑,认为Groesser L等人提出的新的观点对这一类疾病都有启示[9-10]。
引起PPV的基因还有待鉴定。Thomas AC等[11]发现广泛的PPV中激活GNA11和GNAQ突变,基因编码G蛋白的亚基。这些突变在病变组织中检测到的水平非常低,但在血液中检测不到,这也表明这些情况是合子后镶嵌障碍。突变体GNA11(R183C)和GNA11(Q209L)在人细胞系中的体外表达分别激活了下游p38 MAPK信号通路和p38、JNK、ERK通路。在启动子mitfa下表达突变体GNA11(R183C)的转基因斑马鱼嵌合模型出现广泛的真皮黑色素细胞增多,体现了人类的表型。这些发现将允许对这类常见胎记进行准确的临床和分子诊断,从而确诊有严重并发症风险的婴儿,并提供新的治疗机会。
2.3 伴随系统症状:色素血管性斑痣性错构瘤,除了有皮肤的血管和色素沉积异常,还可合并皮肤外症状。PPV患者中约50%的有系统性疾病[12]。常见的“胎记”可能是潜在遗传疾病的一个指标,但往往被忽视。皮肤色素沉积如蒙古斑可以是局部和短暂的,也可以如太田痣是广泛的、永久性的。与血管增生同时发生则定义了一种色素血管增生性疾病的亚型,这是一组与神经、血管、眼科、过度增生和严重并发症相关的综合征。常见合并症有Sturge-Weber综合症、Klippel-Trenaunay综合征、眼球黑变病等。另外散在病例报道合并一些少见症状。Xu等[13]报道1例5个月大的女婴,有广泛的鲜红斑痣和蒙古斑,并有先天性乳糜性腹水。诊断为色素血管性斑痣性错构瘤病合并先天性乳糜性腹水,为Ⅱb型色素血管性斑痣性错构瘤病。认为很可能这两种先天性疾病之间存在联系。Singal等[14]描述1例典型特征为PPV的2个月大的婴儿,合并有明显的腹壁静脉曲张和出生牙,此前未见报道。Shields等[15]认为PPV会增加危及生命的葡萄膜黑色素瘤的风险。有学者报告1例1岁女童PPV患者合并先天性青光眼[16]。Henry[17]报道了1例PPV患者的荧光素血管造影结果,除了先前未描述的视网膜周围血管非灌注的特征外,还发现静脉-静脉吻合。
一旦临床怀疑器官系统受到影响,应进行包括磁共振成像、磁共振血管造影和脑静脉造影、超声心动图、详细的眼科检查、骨骼检查等相关调查。它们有助于确认诊断,并正确描述特定病例的表型特征。
2.4 治疗:色素血管性斑痣性错构瘤病的治疗和预后取决于受影响的器官系统。对于皮肤病变,色素性皮损可以采用Q开关激光、皮秒激光、超脉冲二氧化碳激光、剥脱性点阵激光等。血管畸形可采用脉冲染料激光、长脉宽1 064nm激光或光动力治疗。病例1、病例2色素性皮损经过治疗后颜色基本消退,血管畸形皮损采用脉冲染料治疗,病例2、病例3局部红斑颜色明显减淡。病例1面中部红色增厚处皮损减淡不明显,可能跟此处血管层次较深,血管丰富,血管管径较粗有关,建议采用光动力治疗。病例3中色素性皮损局部治疗后减淡。90%以上黄种人婴儿有蒙古斑损害,大部分病例在5岁前逐渐自然消退,少数可持续到成年期,特别是多发性损害者[18]。本文中病例属于大面积多发色斑患儿,家长希望外观得到改善,笔者科室对此进行小面积尝试性治疗,1次調Q激光治疗后蒙古斑颜色改善明显,提示激光治疗对蒙古斑色素的清除是有效的。考虑到患儿的身体承受能力,暂不考虑大面积治疗。病例4中鲜红斑痣脉冲染料治疗后颜色明显减轻。根据血管畸形的发生部位,皮损厚度,颜色深浅可以初步判断愈后。早期诊断早期治疗,可以减轻疾病对患者社交和心理产生的影响。总之,色素血管性斑痣性错构瘤病需要临床综合评估分型,对于其皮肤改变,在表面麻醉下多种激光联合治疗,可以改善患者面貌,提高生活质量。
[参考文献]
[1]Xu S,Zhang Q,Liu T,et al.A female infant with phacomatosis pigmentovascularis and congenital chylous ascites:A case report[J].Medicine (Baltimore),2018,97(34):12012.
[2]Joshi A,Garg VK,Agrawal S,et al.Port-wine-stain (nevus flammeus), congenital Becker's nevus,café-au-lait-macule and lentigines: phakomatosis pigmentovascularis type Ia-a new combination[J].J Dermatol,1999,26(12):834-836.
[3]黄玉成,李雪莉,李天举,等.色素血管性斑痣性错构瘤病15例临床分析[J].中国皮肤性病学杂志,2012,26(4):315-316.
[4]Happle R.Phacomatosis pigmentovascularis revisited and reclassified[J]. Arch Dermatol,2005,141(3):385-388.
[5]Hasegawa Y,Yauham M.Phakomatosis Pigmentovascularis type IVa[J].Arch Dermatol,1985,121(5):651-655.
[6]Happle R.Dohi memorial lecture,new aspects of cutaneousmosaicism[J].J Dermatol,2002,29(11):681-692.
[7]Happle RMosaicism in human skin,understanding the patterns andmechanisms[J].Arch Dermatol,1993,129(11):1460-1470.
[8]Nanda A,Al-Abdulrazzag HK,Habeeb YK,et al.Phacomatosis pigmentovascularis: Report of four new cases[J].Indian J Dermatol Venereol Leprol,2016,82(3):298-303.
[9]Groesser L,Herschberger E,Sagrera A,et al.Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell[J].J Invest Dermatol,2013,133:1998-2003.
[10]Happle R.Phacomatosis pigmentokeratotica is a “pseudodidymosis”[J].J Invest Dermatol,2013,133:1923-1925.
[11]Thomas AC,Zeng Z,Riviere JB,et al.Mosaic activating mutations in gna11 and gnaq are associated with phakomatosispigmentovascularis and extensive dermal melanocytosis[J].J Invest Dermatol,2016,136(4):770-778.
[12]Kim YC,Park HJ,Cinn YW.Phakornatosis pigmentovasculads type Ⅱa with generalized vitiligo[J].Br J Dermatol,2002,147(5):1028-1029.
[13]Xu S,Zhang Q,Liu T,et al.A female infant with phacomatosis pigmentovascularis and congenital chylous ascites:A case report[J].Medicine (Baltimore),2018,97(34):12012.
[14]Singal A,Mittal H,Aqqarwal A,et al.Phacomatosis pigmentovascularis type 2b (phacomatosis cesioflammea) with double superior vena cava,abdominal varicosities, and natal tooth: Novel associations[J].Pediatr Dermatol,2018,35(3): 151-154.
[15]Shields CL,Di Nicola M,Pelleqrini M,etal.Choroidal melanoma in phakomatosis pigmentovascularis with klippel-trenaunay syndrome[J]. Retina,2018,38(11):2220-2227.
[16]Viada Pelaez MC,Stefano PC,Cirio A,etal. Phakomatosis pigmentovascularis cesioflammea: a case report[J].Arch Argent Pediatr,2018, 116(1):121-124.
[17]Henry CR,Hodapp E,Hess DJ.Fluorescein angiography findings in phacomatosis pigmentovascularis[J]. Ophthalmic Surg Lasers Imaging Retina,2013, 44(2):201-203.
[18]赵辨.中国临床皮肤病学[M].南京:江苏科学技术出版社,2010:1236-1237.
[收稿日期]2019-08-21
本文引用格式:梁文,蒋雨薇,方蔷,等.色素血管性斑痣性错构瘤病4例临床分析及文献复习[J].中国美容医学,2020,29(3):59-62.
猜你喜欢
中国实用医药(2016年13期)2016-07-05
中国实用医药(2016年13期)2016-07-05
上海医药(2016年7期)2016-05-09
中国实用医药(2016年12期)2016-05-04
中国实用医药(2016年10期)2016-05-04
上海医药(2016年3期)2016-03-23
上海医药(2016年1期)2016-02-22
中国实用医药(2016年5期)2016-02-20
现代养生·下半月(2015年11期)2016-01-07
现代养生·下半月(2015年1期)2015-05-26
