
Methane pyrolysis in preparation of pyrolytic carbon: Thermodynamic and kinetic analysis by density functional theory
2020-05-21 04:47ChunxiHUHeSHENShouyngZHANGHejunLI
CHINESE JOURNAL OF AERONAUTICS 2020年3期
Chunxi HU, He SHEN, Shouyng ZHANG, Hejun LI,*
a State Key Laboratory of Solidification Processing, C/C Composites Research Center, Northwestern Polytechnical University,Xi’an 710072, China
b Department of Mechanical Engineering, California State University, Los Angeles, CA 90032, USA
KEYWORDS Density functional theory;Kinetics;Methane pyrolysis;Pyrolytic carbon;Thermodynamics
Abstract The density functional theory has been successfully applied in analyzing pyrolytic carbon deposition by methane pyrolysis from the view of thermodynamics and kinetics based on a total number of 39 elementary reactions. M06-2X/def2-TZVP level was employed to optimize species structures and locate the transition states. The enthalpy changes and Gibbs free energy changes of all the reactions in the temperature range of 298.15-1800 K were derived with optimized species.Results show that the reacting temperature should be above 1200 K based on the equilibrium constant analysis, which is consistent with the typical reaction temperature adopted in experiments.Potential energy surface profiles report that radical attacking reactions have lower energy barriers than those direct decomposition reactions, especially hydrogen radical attacking reactions. The energy barriers of the first steps, dehydrogenations of methane and ethylene, are 272.4 kJ·mol-1 and 288.9 kJ·mol-1 at 1200 K, which are very close to the experimental activation energy for methane pyrolysis.The most favorable decomposition reaction path is the path of hydrogen radical attacking reactions.The highest energy barrier of the path at 1200 K is 185.7 kJ·mol-1 presented by the C-H bond breaking in ethynyl attacked by hydrogen radical.
1. Introduction
Carbon materials are showing a flourishing development with various members, such as carbon nanotubes, graphene, fullerene and composites.1-4Carbon fiber reinforced carbon matrix(C/C) composites are widely used as rocket nozzles and aircraft braking discs5-8in aeronautic industry and further considered as a class of promising materials in electromagnetic interference shielding for sensitive devices,9-11because of their well physical and chemical properties. Indeed, these materials present low density, very good high-temperature mechanical properties, high thermal conductivity and satisfactory electrical conductivity.Generally,such products are elaborated with pyrolytic carbon (PyC) deposited in preformed carbon fibers by hydrocarbon pyrolysis using chemical vapor infiltration(CVI) technique. This process is a topic of great concerns despite it has been widely studied for several years, as its low kinetics and complex transport phenomena still lead to long durations and high costs of fabrications. Formations of PyC with different textures, which have an important influence on the holistic properties of C/C composites,12-15are determined significantly by the kinetic of hydrocarbon pyrolysis.
A series of chemistry and kinetics of methane in CVI were studied in the late 19th century by Benzinger and Hu¨ttinger.The dissociation of methane was assumed to include five gasphase reactions and a surface reaction to form PyC.16To make the assumption more reliable, thermodynamic calculations of gas phase equilibrium were done and more detailed elementary reactions were considered,17following a consecutive reaction scheme:

where, Ci(i=1, 2, 6+, ∞) represents species with i carbon atoms and kj(j=1, 2, 3, 4) is the reaction rate.
Afterward, species concentrations during methane decomposition in preparing PyC were measured by experiments and used to enrich the reaction scheme specifically.18-20In addition, Li and Deutschmann developed a multi-step homogeneous reaction model and a heterogeneous surface reaction to simulate concentration ratios of intermediates in methane pyrolysis and PyC deposition in carbon fiber felts.21Previously, the authors had summarized 909 elementary gas-phase reactions and analyzed reaction pathways of methane in CVI at the molecular level.22With these researches,reaction routes of methane pyrolysis in PyC deposition have been revealed.The resulting kinetic mechanisms were obtained either by summarizing the data in different researches or by using the lumped experimental results and simulated the gas mole fractions and PyC deposition rates in the process,while the bottom of the kinetic modeling, the properties of species and related chemical reactions, is picked with few caution. Liu et al.23studied the polycyclic aromatic hydrocarbon (PAH) radical formations and hydrogen transfer reactions with density functional theory(DFT)and revealed the radical energies and reaction rates in PAH deposition of PyC. Small species are not considered although they are easier to be produced with the initial gas precursor and their depositions are dominant. The species and thermal rate constants of the CH4+H →CH3+H2reaction were accurately calculated by DFT with variational transition state theory and multidimensional semiclassical tunneling state theory24without the discussion of process applications. In the CVI process, PyC deposition is influenced by what kind of carbon precursor is available and how the precursor and its radicals react. The hydrogen atom elimination of carbon precursors forms radicals and initiate a series reactions to grow species and PyC.And the isomerization reactions and the addition reactions to form larger species compete with the dehydrogenation reactions of PyC deposition. Thus, it is substantially important to study the small species energies and their reaction properties in growing PyC for revealing the kinetics of PyC deposition accurately.
In this work,various small species and elementary reactions in methane pyrolysis of PyC preparation are quantified with DFT accurately, aiming at getting the thermodynamic and kinetic properties of the process from the gas precursor to carbon. The reactions are proposed based on bond formations and dissociations among main species,including dehydrogenation reactions, addition reactions and isomerization reactions.Structures of species are first optimized at M06-2X/def2-TZVP level and used in calculations of species thermodynamic parameters (internal energy, enthalpy, Gibbs free energy and entropy) and energy changes of reaction paths.The activation energy of each reaction path is provided by TS method based on the transition state theory. Temperature effects on the above parameters are also studied, revealing the different preferred reaction paths at different temperatures.
2. Reaction pathway design and calculation method
Considering the experimental condition, the industry methane has a purity of 99.5%,the main impurity being ethylene(up to 0.3%).18-20Methane and ethylene, taken as the mother molecules,directly decompose with the breaking of C-H bonds,followed by radical attacking reactions,leading to a huge amount of various radicals, aliphatic hydrocarbons and aromatic hydrocarbons, as well as solid carbon. The present work will only focus on the species with no more than two carbon atoms in order to limit the computer source of high-level calculations in an acceptable time. The simplification of species is reasonable as experimental results have shown that the main species in methane pyrolysis are H2, CH4, C2H2, C2H4, C2H6and C6H6, while the amount of C6H6at a small residence time is zero.18-20Thus several pathways of mother molecule pyrolysis are considered here. Table 1 lists the example reactions of methane and ethylene. More reactions of the radicals and molecules are available in Table A1 in the Appendix A.
DFT at M06-2X/def2-TZVP level was employed to analyze the species and reactions with Gaussian software.25M06-2X is chosen because it has been tested with databases of thermochemistry, kinetics, bond lengths, vibrational frequencies and vibrational zero point energies and is recommended for applications of main-group thermochemistry and kinetics.26-28Frequency analyses were conducted following the structure optimization, which verified the stationary structures (with all real frequencies). Thermodynamic parameters of all structures were performed at different temperatures(298.15-1800.0 K) with zero-point energy corrections. These thermodynamic parameters satisfy29:
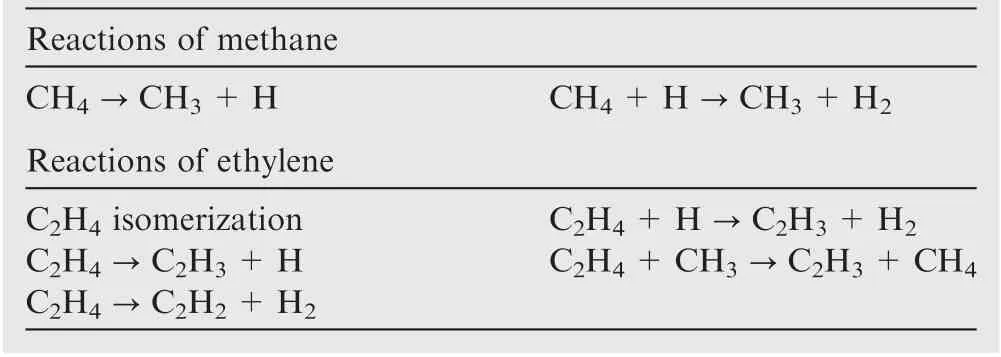
Table 1 Example reactions of methane and ethylene.

Here, E, H, G and S are the internal energy, enthalpy,Gibbs free energy and entropy, respectively. The subscripts 0, elec, zpe, vib, rot, and trans stand for zero-point, electrical,zero-point-correction, vibration, rotation and transition. kBis the Boltzmann constant and T is the temperature in Kelvin.For each reaction path, the energy changes are the energy differences between the reactants and products, since all the atomic information has been cancelled out with the same atom number of each element on both sides of the reactions.29The equilibrium constant K is given by:

where ΔG is the relative Gibbs free energy change and R is the gas constant.
The transition state of each reaction path was located with TS method and confirmed with frequency analysis (a sole imaginary frequency) and intrinsic reaction coordinate (IRC)calculations. The energy analysis of transition states was done at different temperatures,preparing for the kinetic study of the pyrolysis process.
3. Results and discussion
3.1. Structure and vibrational frequencies
The optimization results of structure,symmetry and electronic state for the reactants, products and transition states calculated at M06-2X/def2-TZVP level are shown in Fig. 1 and Fig. B1. Fig. 1 displays structures, symmetries and electronic states of isomers C2H2, C2H3and C2H4and their transition states at M06-2X/def2-TZVP level. Fig. B1 depicts the rest of the species and transition states. No imaginary frequencies are observed in the vibrational frequency analysis for the reactants and products, which confirms that the obtained structures are true minimums on the potential surfaces. Sole imaginary frequency is shown for each transition state. An IRC calculation has been done to determine the reactants and products that linked by the transition state. In Fig. 1,the isomerization reactions expressed with species labels are presented following each transition states. The calculated vibrational frequencies in cm-1of CH4(Td,1A1) are 1348.0,1567.5, 3068.4 and 3188.4 and those for C2H4(D2h,1Ag) are 838.9, 1002.5, 1014.5, 1077.2, 1245.4, 1388.0, 1476.3, 1713.4,3154.5, 3169.5, 3231.8, 3257.9. Compared with the available experimental frequencies30and theoretical results,31the present simulation results are in good agreements,especially when a scale factor (0.97132) is multiplied. The frequencies will be scaled by the factor in the calculations of thermodynamic parameters.
3.2. Thermodynamic analysis of pyrolysis processes
Thermodynamic parameters of each elementary reaction in the pyrolysis process are obtained with its reactants and products.Table 2 lists elementary reactions with any negative enthalpy change ΔH or negative Gibbs free energy change ΔG at different temperatures (298.15, 600, 900, 1200, 1500, 1800 K) at M06-2X/def2-TZVP level. The other reactions with positive ΔH and positive ΔG list in Table C1 in Appendix C, following the sequence of ΔH at room temperature from large to small. For reactions with different structures, their thermodynamic data are calculated separately.Among the total 39 elementary reactions,11 reactions present negative enthalpy changes at all temperatures,indicating an exothermal feature.As listed in the top 11 rows of Table 2 according to the value of ΔH,these reactions are mostly producing stable molecules from active radicals.The values of enthalpy changes in the other reactions are all positive, implying absorbing heat during the reactions. Thus, the mother molecule pyrolysis is generally an endothermic process as most of the reactions show endothermic properties.
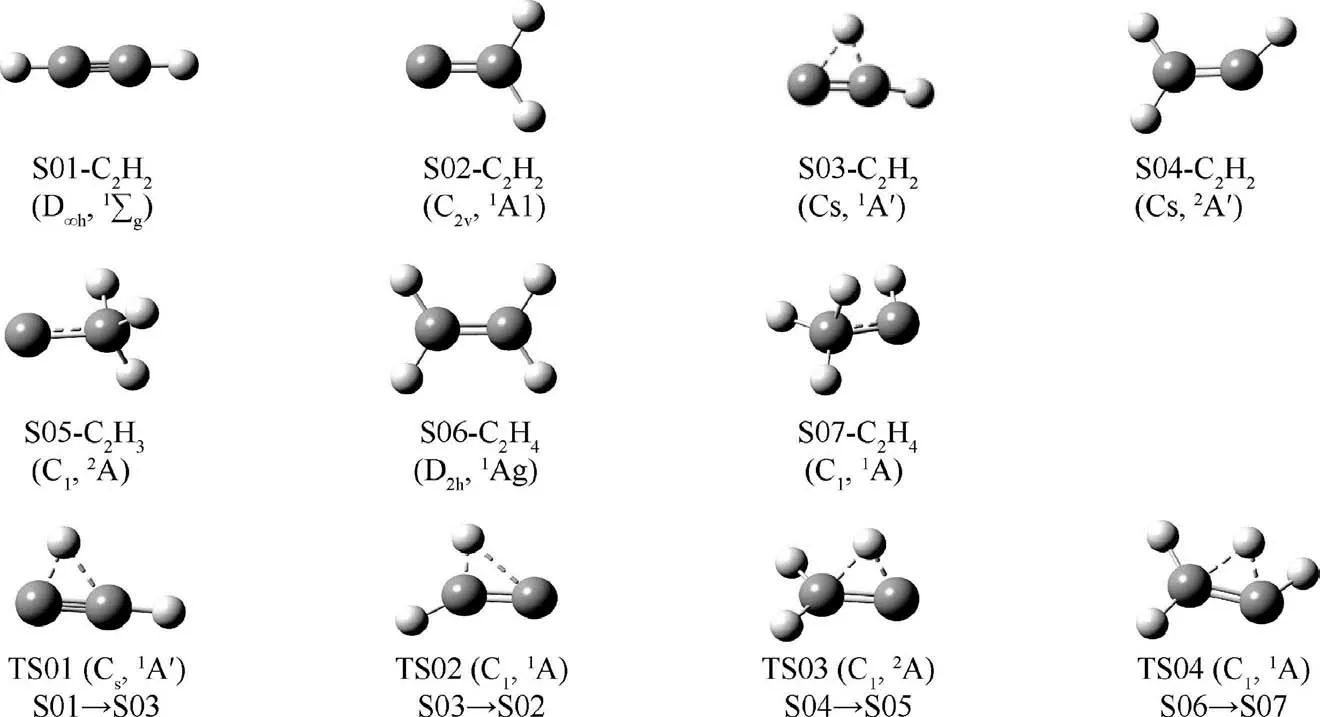
Fig.1 Structures,symmetries and electronic states for isomers C2H2,C2H3 and C2H4 and their transition states at M06-2X/def2-TZVP level.
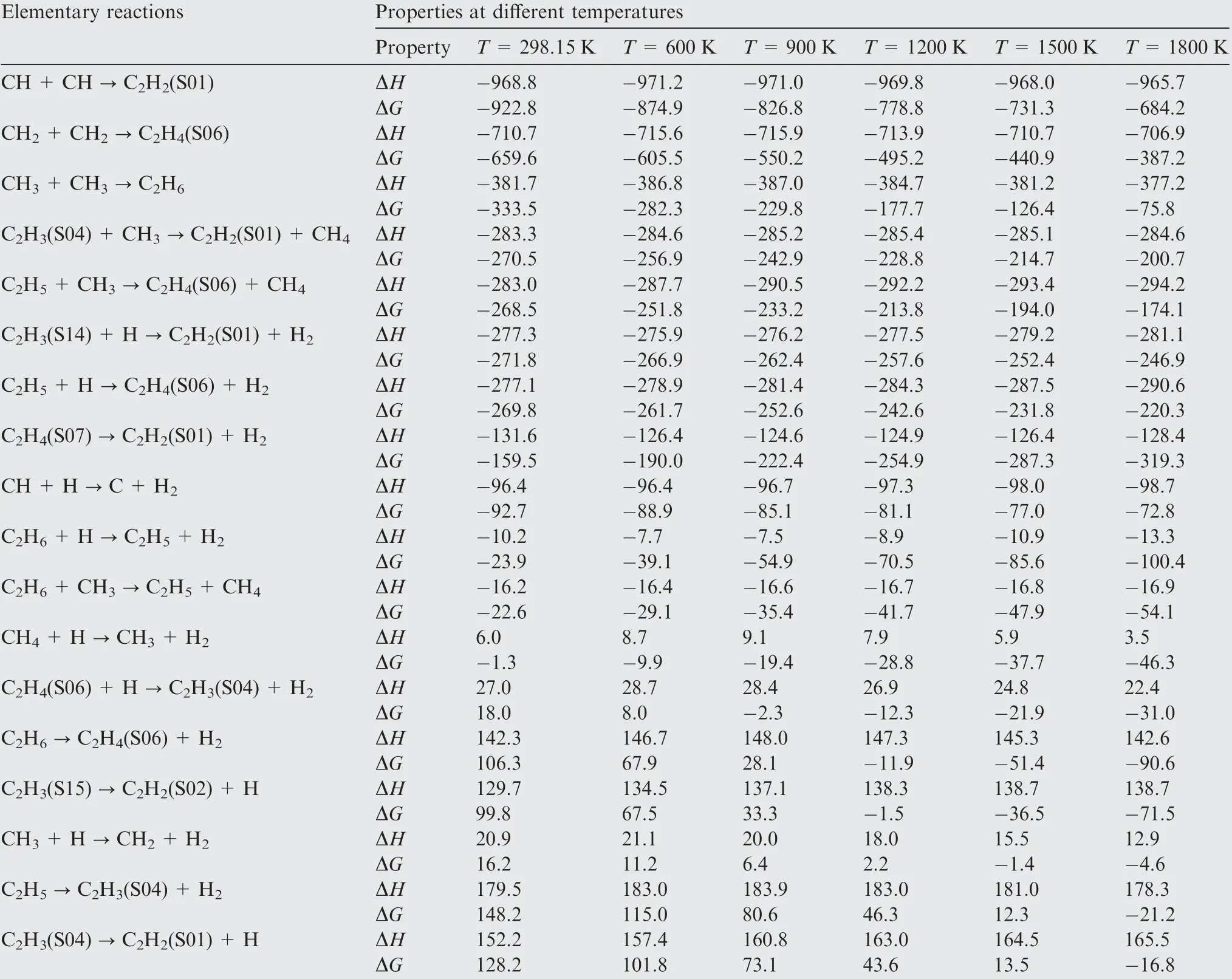
Table 2 Elementary reactions with any negative enthalpy change ΔH or negative Gibbs free energy change ΔG at different temperatures at M06-2X/def2-TZVP level (unit: kJ·mol-1).
The Gibbs free energy change gives the information about the spontaneity of the reaction and the conversion ratio of reactants to products33,34. Reactions with negative ΔG can happen spontaneously.All exothermal reactions show negative values of ΔG above the ambient temperature as shown in Table 2. The endothermic reactions with any negative ΔG are also listed in Table 2 in the order of the temperature at which ΔG becomes negative.The reaction of methane attacked by hydrogen radical can occur spontaneously at room temperature,while the reaction of ethylene attacked by hydrogen radical show negative Gibbs free energies above 900 K.The Gibbs free energy changes of C2H6→C2H4(S06)+H2and C2H3(S05)→C2H2(S03)+H decrease with the increasing temperature and become negative above 1200 K. Similar trends have been shown in the reactions of methyl attacked by hydrogen radical and ethyl cracking into vinyl and hydrogen with the turning points at 1500 K and 1800 K respectively.These show that a high temperature is beneficial for the process.
The conversion ratios of reactions at equilibrium calculated with Eq.(7)give much more visualized information about how much products could be produced. The equilibrium constants increase with the increase of temperature for endothermic reactions and decrease for exothermic reactions.Thus with most of the reaction paths here being endothermic reactions, more products are going to be obtained at high temperatures at the equilibrium condition. At ambient temperature, the equilibrium constants of hydrogen abstraction reactions with methane and ethylene are so small (10-70and 10-74) because of the large Gibbs free energy changes, indicating that the decomposition process is inhibited and the amount of radicals formed is very limited. These equilibrium constants increase dramatically when the temperature increases from room temperature to 1800 K,being 10-6.The values of equilibrium constants for different reactions vary in a very broad range, for example, 10252at 298.15 K with the smallest value being 7.5×10-91(C2H2(S01)→C2H+H) and the largest being 4.7×10-161(CH+CH →C2H2(S01)). Most equilibrium constants show a distinct variation with the increasing temperature and the value range for all reactions narrows quickly.Significant variations of equilibrium constants appear at temperatures below 1200 K. When the temperature increases above 1200 K, the value range of equilibrium constants is no larger than 50, indicating that the process has a quite well conversion.
3.3. Kinetic analysis of pyrolysis processes
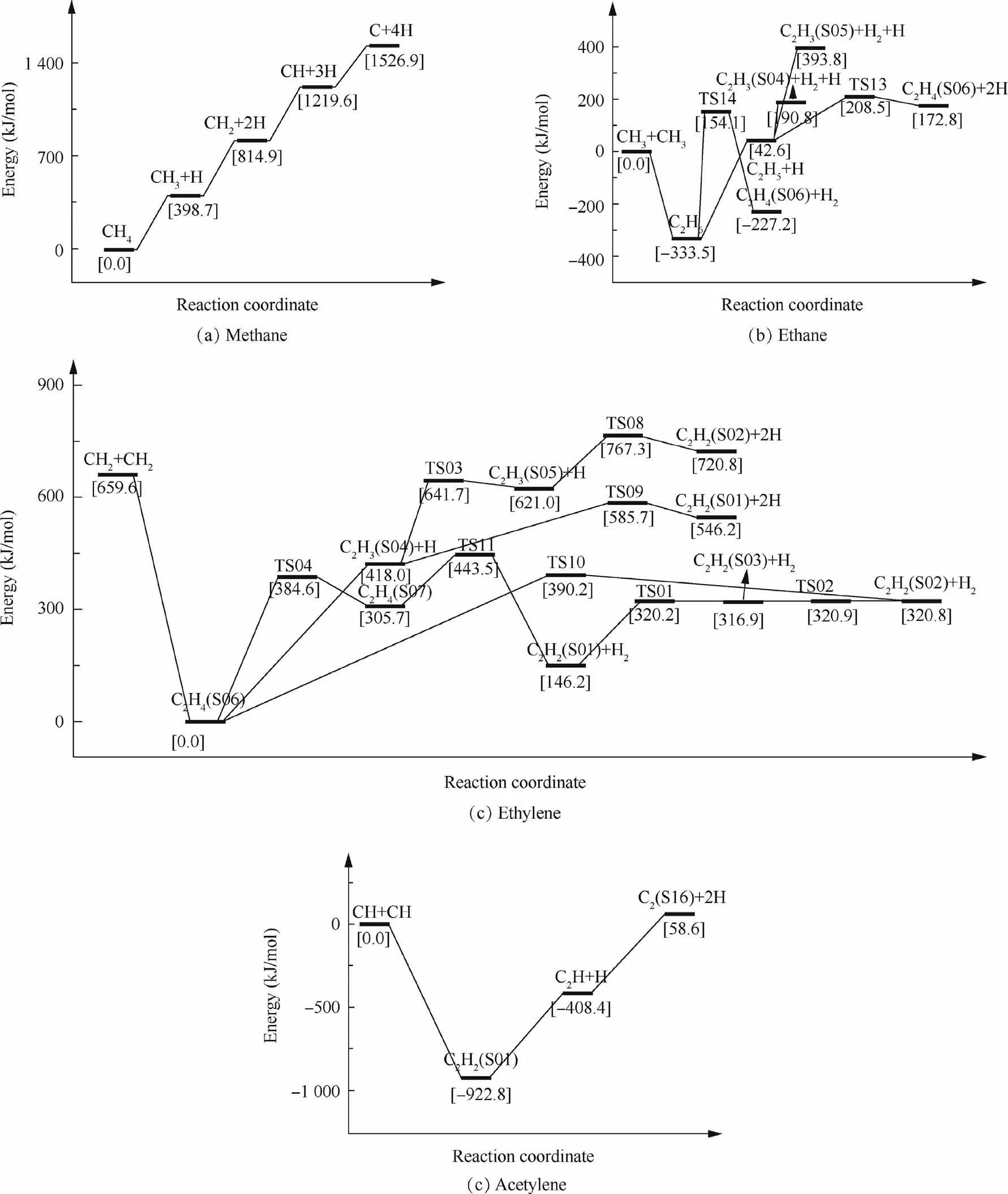
Fig. 2 Potential energy profiles of reactions in methane pyrolysis at room temperature.
The activation energies of the reaction paths are calculated at M06-2X/def2-TZVP level based on the transition state theory.35,36Fig.2 illustrates the potential energy profiles of direct decomposition reaction paths at 298.15 K. Data in the square brackets are relative Gibbs free energies in kJ·mol-1calculated at M06-2X/def2-TZVP level. Fig. 2(a) depicts the direct decomposition of methane. Methane dissociates a hydrogen atom to form methyl with an energy change of 398.7 kJ·mol-1,which excellently agrees with the experimental result.37Following this, one C-H bond is breaking in each step, forming C at last. There are no transition states for these bondcleavage reactions,as no saddle point is found on the potential energy surface. Thus BDEs, that need to be overcome during the reaction process, are approximately considered as activation energies.38The highest energy in the direct methane decomposition is 416.2 kJ·mol-1, for methyl to produce carbene. Fig. 2(b) shows the direct decomposition of ethane.Ethane can be produced by an addition reaction between two methyl radicals and consumed by producing hydrogen radicals or hydrogen molecules. The highest energy barrier is 487.6 kJ·mol-1for the production of ethylene and hydrogen via TS14. Fig. 2(c) presents the direct decomposition of ethylene.Ethylene is either imported as the impurity of the mother gas or produced by an addition reaction of carbene. Ethylene decomposes into vinyl and hydrogen radical with a high energy barrier of 418.0 kJ·mol-1, showing a well consistency with the previous calculation at G3(MP2)//B3PW91 level.39Isomerization of ethylene is much more preferred with a lower energy barrier of 384.6 kJ·mol-1via TS04 for the direct ethylene decomposition, followed by producing acetylene and hydrogen. The isomerization reactions of C2H3and C2H2have also been expressed in Fig. 2(c) and shows that the most stable structures are vinyl and acetylene. Fig. 2(d) is for the direct decomposition of acetylene. Except from the decomposition of ethylene, acetylene can also be formed by the association of two CH radicals. The dehydrogenation reactions give high energy barriers, which are even higher than those in the direct methane decomposition, implying that enough energy should be provided for direct decompositions of carbon deposition.
Apart from the direct decompositions,reactions with active radicals are also available after a few direct decomposition reactions have occurred. Fig. 3 gives potential energy surface profiles of reactions with methyl and hydrogen radicals at 298.15 K.Data in square brackets are relative Gibbs free energies in kJ·mol-1calculated at M06-2X/def2-TZVP level.According to the figure, methyl and hydrogen radicals have lowered the energy barrier in a large amount, indicating easy pathways for decompositions. The energy barrier of methane attacked by hydrogen radical is 77.3 kJ·mol-1, which is only 19.4% of that for the direct methane dehydrogenation. The energy barrier of the dehydrogenation for ethylene attacked by hydrogen radical reduces 79.7% than that of the direct ethylene dehydrogenation. Usually, the reactions with hydrogen radicals express lower energy barriers that those with methyl radicals. The energy barrier for ethane attacked by methyl is 1.3 times as large as that of ethane attacked by hydrogen radical, implying that hydrogen radical is more active than methyl radical.
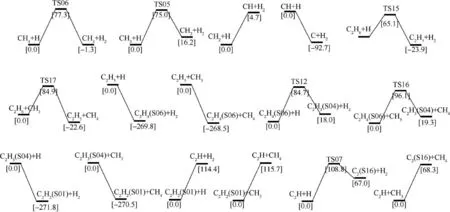
Fig. 3 Potential energy surface profiles of reactions with active radicals at 298.15 K.
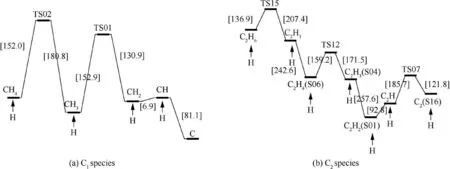
Fig. 4 The most favorable decomposition reaction paths with hydrogen radicals at 1200 K.
With the increase of the temperature, the potential energy profiles vary. The energy barriers of the first C-H bond cleavage in methane and ethylene are 272.4 kJ·mol-1and 288.9 kJ·mol-1at 1200 K, which is very close to the experimental result for methane pyrolysis.17,19For the direct methane decomposition reactions,the highest value of relative Gibbs free energies at 1200 K is 308.0 kJ mol-1for carbene dehydrogenation reaction and decreases 27.7% than that at ambient temperature. The reductions for the energy barriers of direct ethylene and ethane decomposition reactions are 19.1% and 24.1%, given by C2H3(S05) dehydrogenation and ethyl producing C2H3(S05). Ethylene is preferred to produce vinyl and hydrogen radical at 1200 K, which is quite different from the situation at 298.15 K. In direct acetylene decomposition reactions, the relative energy of the final product C2(S16)compared with that of acetylene at 1200 K is much lower than that at room temperature, which expresses a more stable state at high temperatures. While the radical attacking reactions show increasing energy barriers with the increase of the temperature, reactions with radicals are still more favorable than the direct decomposition reactions and the hydrogen radical is still more active than methyl at 1200 K. Fig. 4 depicts the most favorable decomposition reaction paths with hydrogen radicals at 1200 K. Data in square brackets along the paths are relative Gibbs free energies in kJ·mol-1calculated at M06-2X/def2-TZVP level. Methane is attacked by hydrogen radical to produce methyl via TS06 with an energy barrier of 152.0 kJ·mol-1. Then one C-H bond of methyl could be broken by hydrogen radical attacking via TS05 with an energy barrier of 152.9 kJ·mol-1, showing the highest energy barrier in C1decomposition (Fig. 4(a)). There is only a small energy difference between the reactants and products in CH2+H →CH+H2,being 6.9 kJ·mol-1.To release C from CH, the energy change is -81.1 kJ·mol-1, making C the most stable species among all the C1species. Fig. 4(b) shows the most favorable decomposition reaction paths of C2species with hydrogen radicals at 1200 K. Ethane attacked by hydrogen radical forms ethyl via TS16 with an energy barrier of 136.9 kJ·mol-1. An energy decrease is found in ethylene produced from vinyl attacked by hydrogen radical. Ethylene attacked by hydrogen radical could dissociate a hydrogen atom to form vinyl via TS12 with an energy barrier of 159.2 kJ·mol-1. The most stable species in the C2decomposition process is acetylene, producing from vinyl attacked by hydrogen radical with an energy reduction of 257.6 kJ·mol-1.Acetylene is attacked by hydrogen radical and forms C2H,showing an increasing energy of 92.8 kJ·mol-1. C2H decompose the last hydrogen atom to form C2(S16) via TS07 with the highest energy barrier of 185.7 kJ·mol-1in the decomposition process.
4. Conclusions
(1) Methane decomposition in preparation of pyrolytic carbon was studied by an accurate chemistry model based on the density functional theory with a total number of 39 elementary reactions.Stable species and transition states were optimized at M06-2X/def2-TZVP level.Molecular energies in the temperature range of 298.15-1800 K were calculated with the optimized structures and scaled frequencies and used in the calculations of enthalpy changes and Gibbs free energy changes of reactions.
(2) Most of the enthalpy changes are positive, implying the endothermic feature of the pyrolysis process.Analysis of Gibbs free energy changes show that all the exothermic reactions can happen spontaneously at room temperature, as well as the reaction of methane attacked by hydrogen radical. Well conversion ratios are obtained for all reactions above 1200 K, consistent with the typical reaction temperature employed in experiments.
(3) Potential energy surface profiles of the pyrolysis process including direct decompositions and radical attacking reactions at ambient temperature show that radical attacking reactions have lower energy barriers than those direct decomposition reactions, especially hydrogen radical attacking reactions. As the temperature increases, the energy barriers of direct decomposition reactions decrease.
(4) The first steps C-H bond cleavages in methane and ethylene present energy barriers of 272.4 kJ·mol-1and 288.9 kJ·mol-1at 1200 K, which is very close to the experimental activation energy for methane pyrolysis.The most favorable decomposition reaction paths with hydrogen radicals at 1200 K were illustrated with the highest energy barrier of 185.7 kJ·mol-1for the C-H bond breaking in ethynyl attacked by hydrogen radical.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51821091 and 51472203), the ‘‘111”Project of China(Grant No.B08040),and the Research Fund of State Key Laboratory of Solidification Processing(NWPU)of China (Grant No. 142-TZ-2016).
Appendix A. Reactions

Table A1 Reactions of radicals and molecules except those with methane and ethylene.
CH3→CH2+H CH2+CH2→C2H4
CH3+H →CH2+H2CH →C+H
Table A1 (continued)
Appendix B. Species and transition states
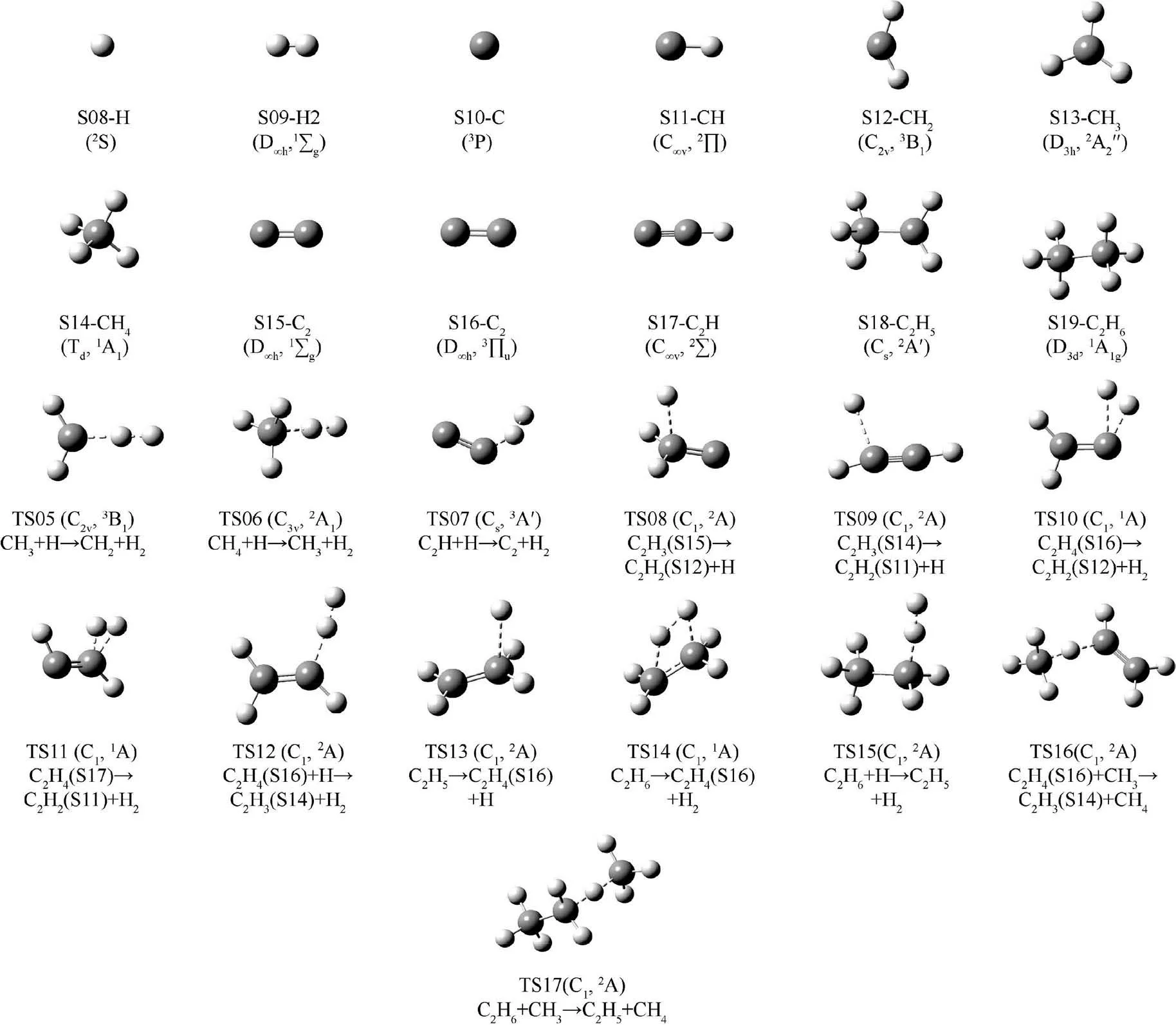
Fig. B1 Structures, symmetries and electronic states for stable species without isomers and transition states of dehydrogenation reactions at M06-2X/def2-TZVP level.
Appendix C. Enthalpy changes and Gibbs free energy changes
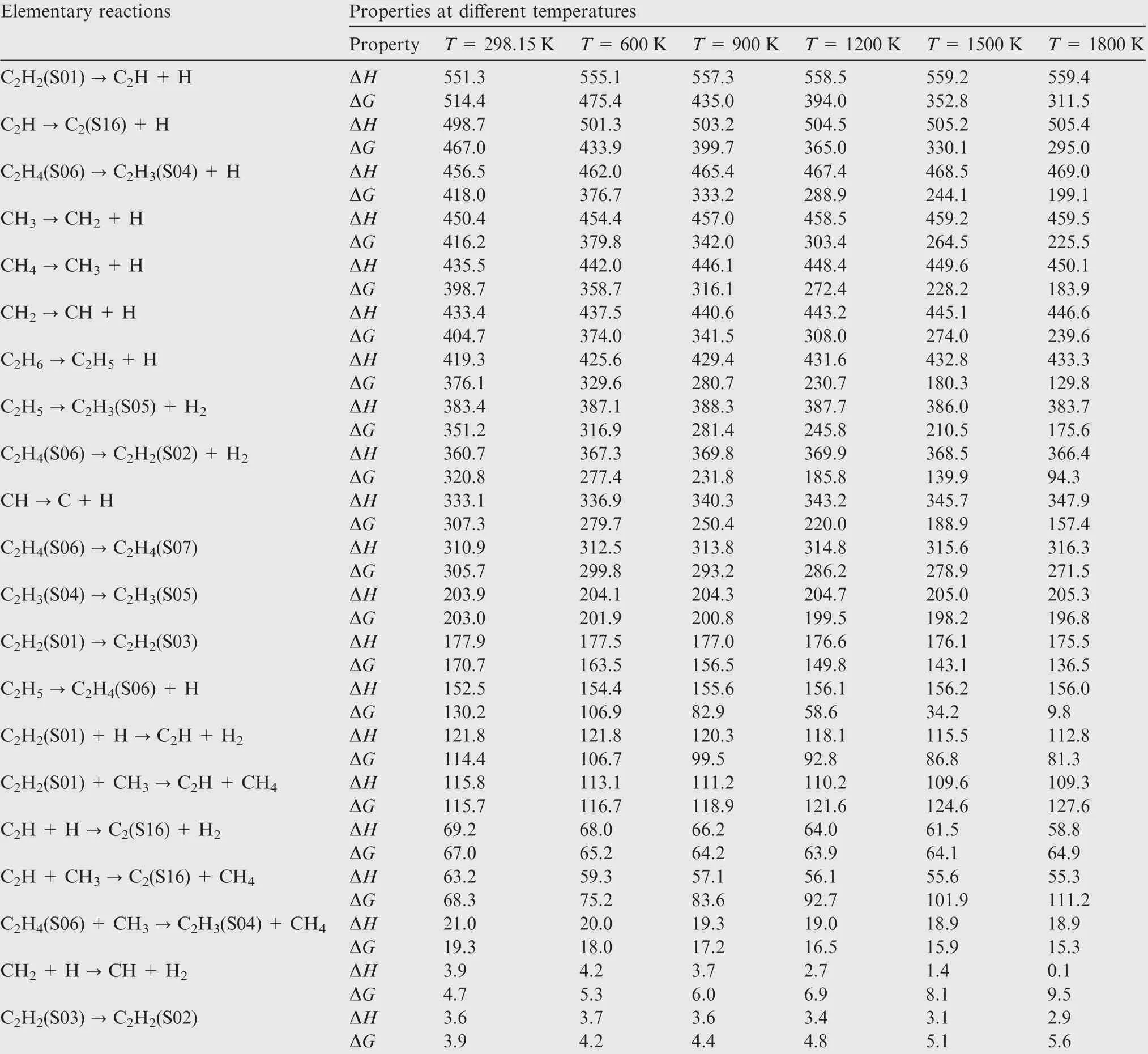
Table C1 Elementary reactions and their positive enthalpy changes ΔH and positive Gibbs free energy changes ΔG at different temperatures at M06-2X/def2-TZVP level (unit: kJ mol-1).
 CHINESE JOURNAL OF AERONAUTICS2020年3期
CHINESE JOURNAL OF AERONAUTICS2020年3期
- CHINESE JOURNAL OF AERONAUTICS的其它文章
- Experimental investigation on operating behaviors of loop heat pipe with thermoelectric cooler under acceleration conditions
- Investigation of hot jet on active control of oblique detonation waves
- Experimental study of rotor blades vibration and noise in multistage high pressure compressor and their relevance
- Unsteady wakes-secondary flow interactions in a high-lift low-pressure turbine cascade
- Effect of protrusion amount on gas ingestion of radial rim seal
- Optimization design of chiral hexagonal honeycombs with prescribed elastic properties under large deformation