
Nephrocystin-3基因突变致婴儿型肾单位肾痨一例
2020-05-13 12:41翟春桃匡新宇孙蕾孙利文王平吴滢黄文彦
临床肾脏病杂志 2020年3期
翟春桃 匡新宇 孙蕾 孙利文 王平 吴滢 黄文彦
200062 上海,上海交通大学附属儿童医院肾脏风湿科(翟春桃,匡新宇,孙蕾,孙利文,王平,吴滢,黄文彦);470000 洛阳,洛阳市妇幼保健院儿科(翟春桃)
病 例 资 料
患者,女,1岁9个月。因“皮肤瘙痒3个月,发现血肌酐升高1个月”于2019年4月9日入住我科。患儿于入院前3个月无明显诱因出现全身皮肤瘙痒,无发热、咳涕,无吐泻,无皮疹、关节疼痛,无浮肿等不适;至当地医院皮肤科间断治疗2个月,症状无改善。20余天前无明显诱因下出现呕吐、腹泻,呕吐非喷射性,呕吐物无血丝、血块,解稀水便5~6次/d。至当地医院就诊,查腹部超声示双肾实质回声增强;遂于2019年3月14日至2019年4月10日于某专科医院住院治疗,查粪常规轮状病毒弱阳性,动脉血气分析检测:pH值7.2,二氧化碳分压(PCO2)19.7 mmHg,氧分压(PO2) 138.0 mmHg,剩余碱(BE(B))-18.5 mmol/L;外周血常规:白细胞4.71×109/L,血红蛋白63 g/L,血小板226×109/L;血生化:丙氨酸转氨酶(ALT)251 U/L,天冬氨酸转氨酶(AST) 126 U/L,碱性磷酸酶(AKP)841 U/L ,肌酐(Cr)139 μmol/L,尿素氮(BUN)19.54 mmol/L,尿酸(UA)555 μmol/L,总胆固醇(TC)9.1 mmol/L,降钙素原(PCT) 1.42 ng/ml,尿常规示pH 7.0,尿蛋白阴性,红细胞0~1/HP,白细胞0~1/HP,凝血功能、补体C3、补体C4正常,泌尿系统超声示双肾增大(左7.6 cm×3.6 cm;右7.4 cm×2.7 cm)、双肾弥漫性病变,腹部超声、胸片未见明显异常,肾穿活检光镜示小球发育幼稚伴部分硬化及明显间质小管病变;电镜示肾小球基底膜明显皱缩,足突大部分融合,有微绒毛变,未见确切电子致密物沉积,给予补液、纠酸、保肝、降压等对症治疗,4月2日(入院后第20天)复查生化示血ALT 410 U/L, AST374 U/L,血Cr178 μmol/L,估算肾小球滤过率(eGFR) 21.4 mL·min-1·(1.73 m2)-1,建议完善肝脏穿刺活检,家属拒绝。于2019年4月14日转至我科治疗。
既往史:患儿于2018年10月因“肠套叠”在当地行手术治疗,当时有镜下血尿及双肾弥漫性改变,但查SCr47 μmol/L,ALT43 U/L,未予特殊处理。
个人史:患儿系G2P2,生后无窒息史,母乳喂养,精神、运动发育与同龄儿无异,按时预防接种,无传染病接触史,母亲孕期孕检无异常。
家族史:患儿父母均有肾结石病史,其姐姐身体健康。否认家族性遗传病史,否认家族成员肾脏病史。
体格检查:体温 36.4 ℃,心率101次/min,身高78 cm,体质量10 kg,血压127/79 mmHg。神清,反应可,营养状况一般,身高P3-P10,体重P3,贫血貌,全身皮肤及粘膜无出血点,无水肿及黄染,双侧颈部未触及肿大淋巴结,咽无充血,双肺未闻及干湿性啰音,心音有力,心律齐,各瓣膜区未闻及明显杂音,腹软,无压痛、反跳痛,右下腹见8 cm陈旧性瘢痕组织,四肢活动可,肌力、肌张力正常,双下肢无水肿,关节无肿胀,神经系统:生理性反射存在,病理性反射未引出。
入院后辅助检查、诊断及治疗经过如下。
患儿入院后完善相关检查。尿液分析:尿蛋白多次检查为阴性或弱阳性,24 h尿蛋白定量因患儿不配合难以留取,血白蛋白持续>40 g/L,血肌酐进行性升高,从77 μmol/L升高至195 μmol/L,且患儿有贫血,血红蛋白为63 g/l,代谢性酸中毒(血气分析pH值7.29,PCO24.40 Kpa,BE(B) -9.80 mmol/L,HCO315.90 mmol/L),泌尿系统超声提示双肾增大、双肾弥漫性病变。结合外院肾穿刺活检示小球发育幼稚伴部分硬化及明显间质小管病变,电镜示肾小球基底膜明显皱缩,足突大部分融合,有微绒毛变,未见确切电子致密物沉积;入院后测算eGFR<15 mL·min-1·(1.73 m2)-1,慢性肾脏病5期(CKD5)诊断明确。住院期间评估各项并发症:(1)生长发育与营养:患儿身高位于同年龄同性别儿童P3-P10,体重P3;(2)心血管:患儿入院后监测血压,平均压115/80 mmHg,心超提示二尖瓣轻微反流;(3)血液系统:患儿外周血血红蛋白波动于54~98 g/L,血清铁6.81 μmol/L,铁蛋白177.8 ng/ml,促红细胞生成素5.00 MIU/ML;(4)钙磷代谢:钙正常,磷稍高,25羟维生素D(123.30 nmol/L)充足;(5)肝功能不全:入院后检测肝功能示ALT251 U/L, AST126 U/L,AKP841 U/L。考虑患儿处于CKD5期并伴贫血、肝功能不全、心血管系统损害等并发症,行腹膜透析肾脏替代治疗,腹透前间断予连续肾脏替代疗法(continuous renal replacement therapy,CRRT)治疗,严格低盐低蛋白饮食,控制液体摄入量,予保肝、降压、纠酸、纠正贫血等对症治疗,CRRT后血肌酐可明显下降。住院期间全外显子基因检测报告回示:本次样本检测发现受检者基因组Nephronophthisis(NPHP)3 基因 c.3870_3873del 位点和 c.A3442T 位点发生复合杂合型变异,其父母均为NPHP3杂合子,正常无表型,受检者在上述两位点发生复合杂合型变异。(图A、图B)
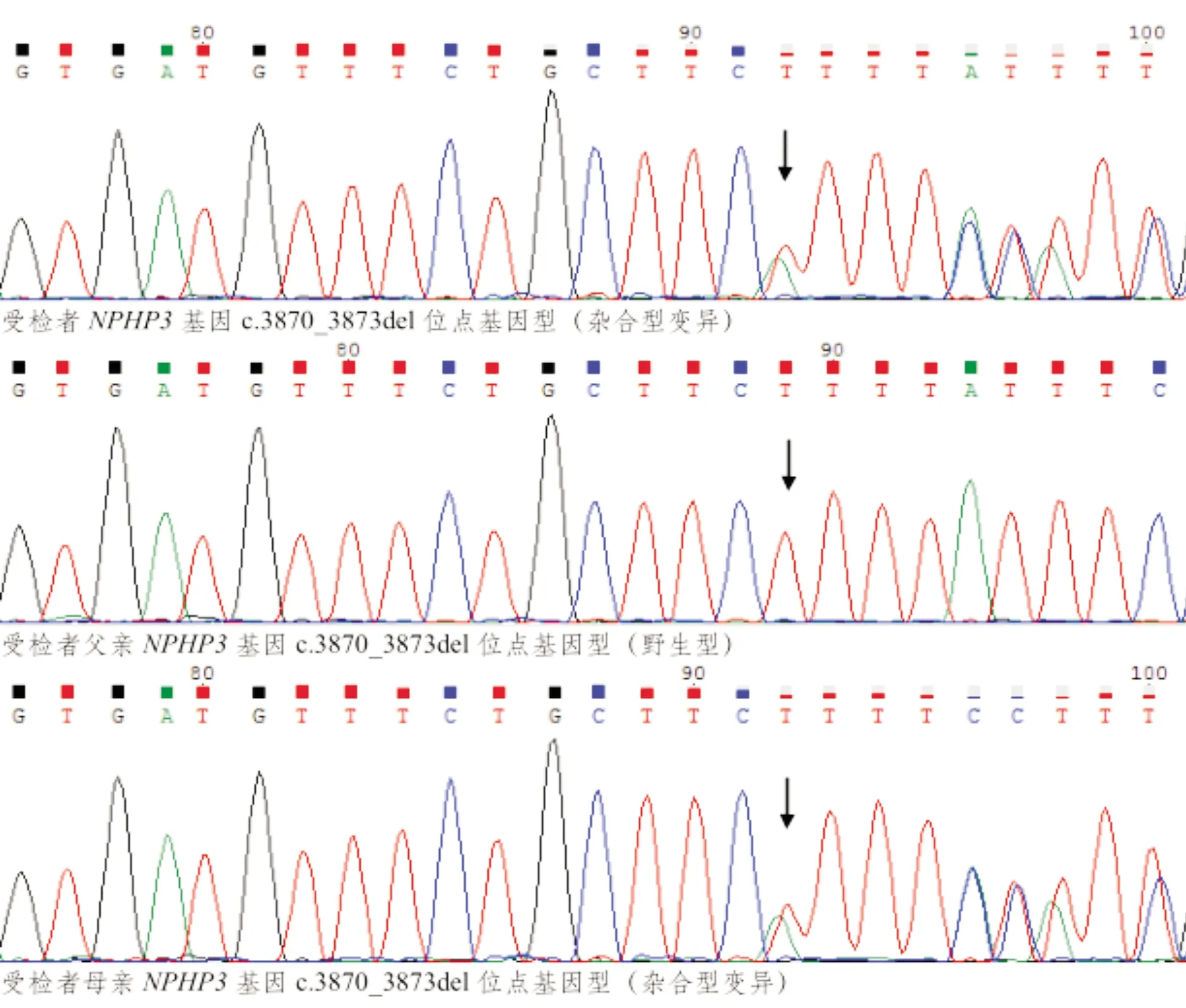
图1A患儿NPHP3基因c.3870_3873del位点家系验证图
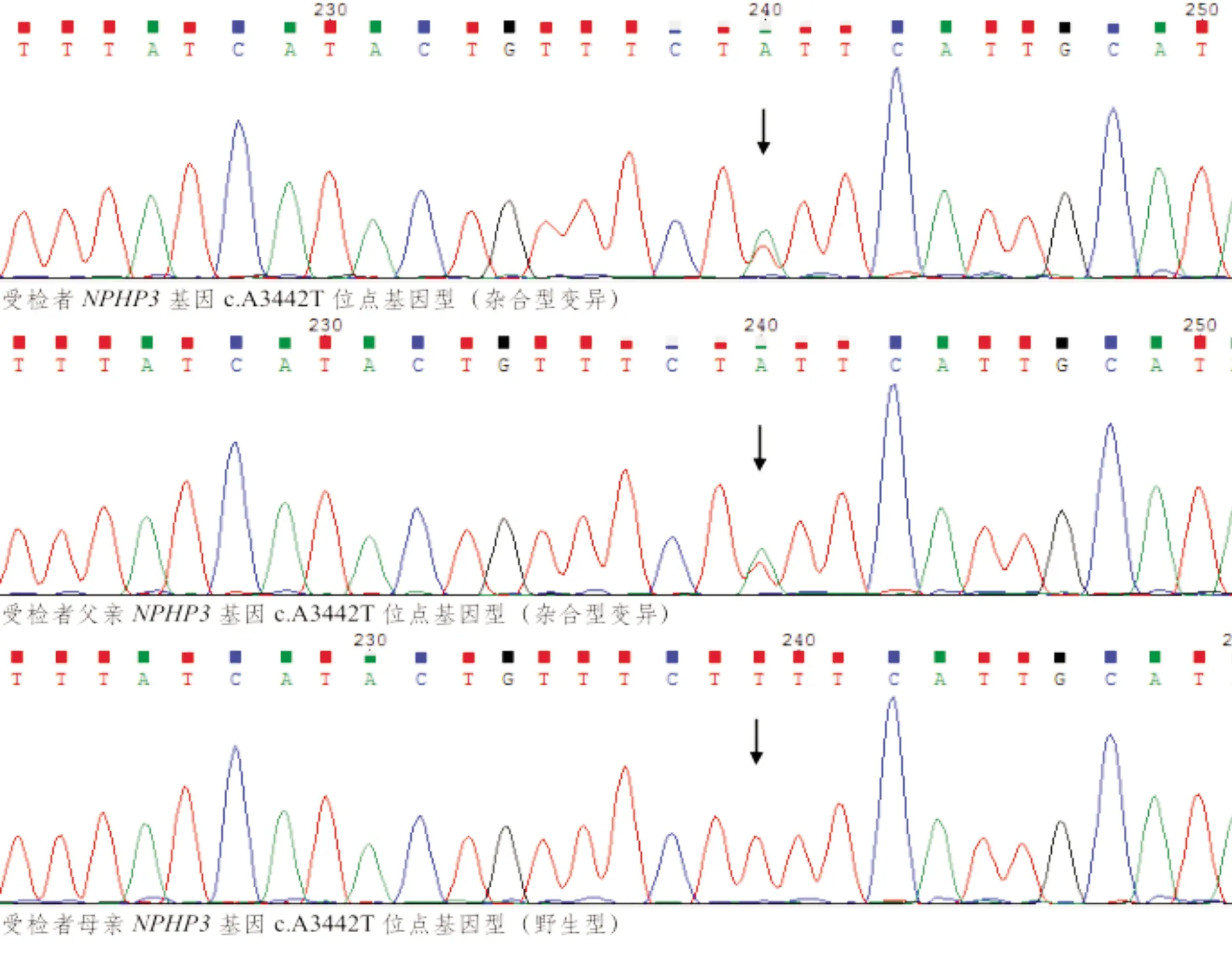
图1B患儿NPHP3基因c.A3442T位点家系验证图
讨 论
本文报道1例21月龄的女性患者,以皮肤瘙痒起病,病程中伴有肝功能损害,贫血,高血压,尿酸增高,血肌酐进行性升高,起病隐匿,半年内快速进展至终末期肾脏病(ESRD)。B超提示双侧肾脏增大、弥漫性病变,肾脏穿刺活检示肾小球发育幼稚伴部分硬化及明显间质小管病变;电镜示肾小球基底膜明显皱缩,足突大部分融合,有微绒毛变,未见确切电子致密物沉积;结合患儿无蛋白尿、肾组织活检提示肾小管间质病变,高度考虑遗传性因素所致,给予全外显子基因检测(靶向捕获+高通量测序)明确存在NPHP3基因变异,该基因 c.3870-3873del 位点和 c.A3442T 位点发生复合杂合型变异,分别来源于母亲、父亲。受检者(系本文患儿) NPHP3 基因在 chr3:132400873 位置上发生碱基CTTTT>C 的杂合缺失变异,导致编码的第 1290 为氨基酸后发生移码变异,按ACMG 序列变异分级标准判断为临床意义未明(证据:PM2)。在 chr3:132403526 位置上发生碱基A>T 的杂合变异,该位点为无义变异,按 ACMG分级标准判断为致病性(证据:PSV1,PM2,PP3)。上述两个位点变异在千人基因组、ExAC 以及 gnomAD 等数据库中均未被收录。
肾单位肾痨是一种主要累及肾小管间质的常染色体隐性遗传的囊性肾脏病,随病程进展可快速进入ESRD,根据其ESRD发病的中位年龄,可分为婴儿型(2岁前)、少年型(平均年龄为13岁)、青年型(平均年龄为19岁)[3],这些类型与特定的基因缺陷有关。发病相对罕见,无性别倾向,关于其发病率报道有地区差异,加拿大1∶50 000,芬兰1∶61 800,美国1∶1 000 000[4],我国尚无其发病率报道,但经基因检测明确NPHP诊断的至今共报道20余例。
NPHP的临床表现复杂多样,起病隐匿,大部分患儿以多饮、多尿、贫血、生长发育迟缓等就诊,随疾病进展逐渐出现肾功能减退。其影像学无特殊表现,李国民等[5]总结了目前国内外新生儿期进展为ESRD的NPHP患儿,这些患儿在胎儿期超声提示羊水少、双肾增大伴囊性改变,易被误诊为常染色体隐性遗传多囊肾。婴儿型NPHP由于起病年龄小,肾功能恶化迅速,在影像学方面可表现为疾病早期双肾体积增大,后期缩小;少年型和青年型无特殊表现。NPHP患儿肾脏病理典型表现为:光镜下为肾小管间质病变三联征,即肾小管基底膜完整性破坏,表现为不规则增厚或变薄;小管萎缩和髓质囊性变;肾脏间质单个核细胞浸润和纤维化。肾小球病变早期轻微,晚期可出现继发的节段性肾小球硬化。电镜下肾小管基底膜有明显增厚,也可见基底膜断裂[6]。婴儿型肾脏病理缺乏典型的肾小管基底膜改变,且在皮髓交界处可见囊肿形成[7]。结合本例患儿,其临床表现并不典型,以无明显诱因皮肤瘙痒起病,但仔细回顾患儿病史,可发现其生长发育迟缓,与同龄患儿相比,身高、体重均位于P3,伴有贫血、肝功能不全,起病后迅速进展至ESRD,B超提示双肾体积增大,肾穿刺活检明确其病变部位在肾小管间质,全外显子基因检测明确存在NPHP3基因复合杂合变异,在chr3:132403526 位置上发生碱基A>T的杂合变异,该位点为无义变异、有害性突变。故肾脏病理、基因检测在NPHP病患的诊断中显得尤为重要。
NPHP发病的分子机制并不明确,患者存在纤毛蛋白编码基因的突变,蛋白质产物表达于原纤毛、基体和中心体,通常被称为与纤毛相关性疾病,而纤毛相关基因的缺失又可引起一系列疾病,包括NPHP、Joubert 综合征、Meckel-Gruber 综合征和Bardet Biedl 综合征等[8-9]。目前已经明确的与NPHP有关的致病基因有20个,其基因编码蛋白被称为nephrocystins[10]。nephrocystins表达于多种组织的初级纤毛,但不是均匀的分布于纤毛内,它分享共同的蛋白组件,干涉、调节多个信号传导通路,可调控器官形态的发育和维持其功能作用[10-12,6]。NPHP3位于3q22,编码一种有微管蛋白-酪氨酸连接酶域的蛋白,其与nephrocystin-1蛋白相互作用,具有27个外显子[13],是一种含有1325个氨基酸的蛋白质,具有3个n端螺旋结构域和8个四肽重复序列[14]。NPHP3突变方式主要是点突变,包括单个或多个碱基的缺失、替换或插入[10]。国外有研究认为NPHP3参与调节非经典Wnt通路,其中任何一种功能受损都可能破坏平衡,导致发育不良,从而表现出相关的临床症状[13]。日本有学者[15]认为NPHP3与INV协同调控肾纤毛ANKS6磷酸化,并且作用于ANKS6上游,他们的研究证实了一种新的纤毛信号通路ANKS6作为一个信号中介来连接纤毛和细胞质,从而调控肾脏形态的发生。
NPHP3所致婴儿型肾单位肾痨的临床病例报道甚少,Otto等[16]在2例婴儿型肾单位肾痨患者中检测到其突变。Tory等[17]在对5岁以内的43个家系婴儿型NPHP的研究中发现NPHP3的突变率为16%。孙良忠等[18]对15例5岁内婴儿型NPHP的检测,NPHP3突变率53.3%。李国民等[5]在2017年报道了2例婴儿型NPHP,均为NPHP3的复合杂合突变。由于本病起病隐匿,故对其<5岁不明原因出现ESRD的患儿,均应高度警惕该病可能,尽早行基因检测,同时需警惕其他儿童囊性肾脏疾病,如髓质囊性病、髓质海绵肾等。
本例患儿在起病半年内快速进展至ESRD,鉴于其年龄因素,行腹膜透析治疗,腹透前间断予CRRT治疗,患儿病情稳定后出院门诊随访。NPHP病患的治疗目前尚无统一治疗方案,治疗原则与CKD相同,主要是对症,延缓肾衰竭的发生,进展至终末期的患儿给予CRRT治疗。肾移植或肝肾联合移植或许是目前发展至ESRD患儿的一个比较好的选择,可改善其生活质量和生存率。NPHP是常染色体隐性遗传病,除显著肾小管间质损伤外,临床表现复杂多样,提高对其认识,加强相关家系或疑诊患儿基因检测可防止误诊、漏诊。
猜你喜欢
医学综述(2021年18期)2021-12-03
医学研究生学报(2021年4期)2021-12-02
中国科技教育(2021年1期)2021-06-11
辐射研究与辐射工艺学报(2021年2期)2021-05-06
种子(2021年3期)2021-04-12
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
滨州医学院学报(2016年2期)2016-05-27
