
肾活检发现Fabry病一例分析
2020-05-13 12:41戴秀娟吴杰吕小萌王小龙聂飒飒
临床肾脏病杂志 2020年3期
戴秀娟 吴杰 吕小萌 王小龙 聂飒飒
363000 漳州,福建省漳州市中医院糖肾科(戴秀娟);100853 北京,中国人民解放军总医院第一医学中心肾脏病科(吴杰,吕小萌,王小龙,聂飒飒)
病 例 资 料
患者,男性,28岁,因“发现尿检异常1个月”于2018年11月12日收住解放军总医院肾脏病科。患者于2018年10月进食阿根廷红虾及药酒后出现发热,自测体温38.2℃,伴颜面部及双眼睑浮肿,泡沫尿,双下肢无水肿,不伴少尿、肉眼血尿、皮疹、关节痛、光过敏等症状,口服氯雷他定、对乙酰氨基酚及连花清瘟胶囊后症状缓解。院外查尿红细胞25/μL、24h尿蛋白定量0.867 g、尿微量白蛋白/肌酐比值>144 mg/g,为行肾穿刺活检入院。既往史:平素进食高热量食物后出现手足灼热或感觉异常。家族史:父亲有肾结石及高血压病史,查尿常规正常;母亲体健。有同母异父的1姐31岁和1弟25岁,均体健。入院查体:体温36.5℃,脉搏79次/分,呼吸18次/分,血压102/68 mmHg,体质量指数18.3。体格检查未见明显异常。
辅助检查:血清白蛋白35.8 g/L,血肌酐58.2 μmol/L。尿常规:尿比重1.015,尿红细胞0~3/HPF,尿蛋白70 mg/dL。尿红细胞形态均一、计数2个/μL。尿液N-乙酰-β-D-葡萄糖苷酶10.5 U/L、尿渗透压540 mOsm/L。补体:C3 89.2 mg/dL,C4 18.8 mg/dL。免疫球蛋白:IgA 306 mg/dL,IgE 1100 IU/mL,IgG、IgM、κ轻链、λ轻链未见异常。尿微量白蛋白48.3 mg/dL、尿液IgG 3.11 mg/dL、尿液Ig轻链κ1.53 mg/dL、尿液Ig轻链λ 0.828 mg/dL、尿转铁蛋白3.0 mg/dL。血沉 8 mm/h,抗“O”<200 U/ml。抗核抗体谱、抗磷脂酶A2受体抗体、抗肾小球基底膜抗体、抗中性粒细胞胞浆抗体均阴性。24h尿蛋白定量:1.64 g。心电图:窦性心律,左心室高电压。泌尿系超声:左肾囊肿。胸部CT平扫、超声心动图、双肾静脉+肝胆胰脾+腹腔超声均未见明显异常。眼科检查:双眼底未见明显异常,未见角膜异常,考虑患者进食红虾和药酒后起病,不能除外急性间质性肾炎,入院后给予口服醋酸泼尼松片30 mg、1次/日抗炎、抑制免疫,氯沙坦钾50 mg、1次/日及黄葵胶囊降尿蛋白治疗。
肾脏病理报告:光镜:36个肾小球,肾小球体积增大,6个全球硬化(17%),足细胞广泛空泡样变,部分毛细血管袢皱缩、开放不良,壁层上皮细胞节段增殖,包曼氏囊壁节段增厚。肾小管见灶性、轻度萎缩,部分上皮细胞空泡样变,管腔内偶见蛋白管型。间质见灶性、轻度炎细胞浸润,炎细胞以淋巴/单核细胞为主,见灶性、轻度纤维化,见图1。免疫荧光(3个肾小球):IgG(-)、IgA(-)、IgM(-)、C3(-)、C4(-)、C1q(-)、纤维蛋白原(-),考虑足细胞病,建议送电镜排除Fabry病。根据光镜病理结果给予停用糖皮质激素。电镜:肾小球毛细血管内皮细胞明显空泡变性,个别管腔内可见红细胞。肾小囊壁层细胞空泡变性,脏层上皮细胞肿胀、空泡变性,呈泡沫状,次级溶酶体增多并见髓样小体和斑马小体,足突弥漫融合。1个系膜区可见微管样结构,直径约30 nm。未见电子致密物沉积。肾小管上皮细胞空泡变性。肾间质无特殊病变。病理诊断:符合Fabry病肾病,见图2。基因检测:GLA基因的EX6/CDS6检出c.973G>C突变,氨基酸变化为p.Gly325Arg;染色体位置chrX:100653384,为半合子错义突变;变异类型:临床意义未明,见图3~4。血β-半乳糖苷酶活性检测(比色法):151.8 nmol/L(正常范围>90)。患者口服氯沙坦及黄葵胶囊治疗2个月后随访:24h尿蛋白定量0.597 g,血肌酐56.5 μmol/L。
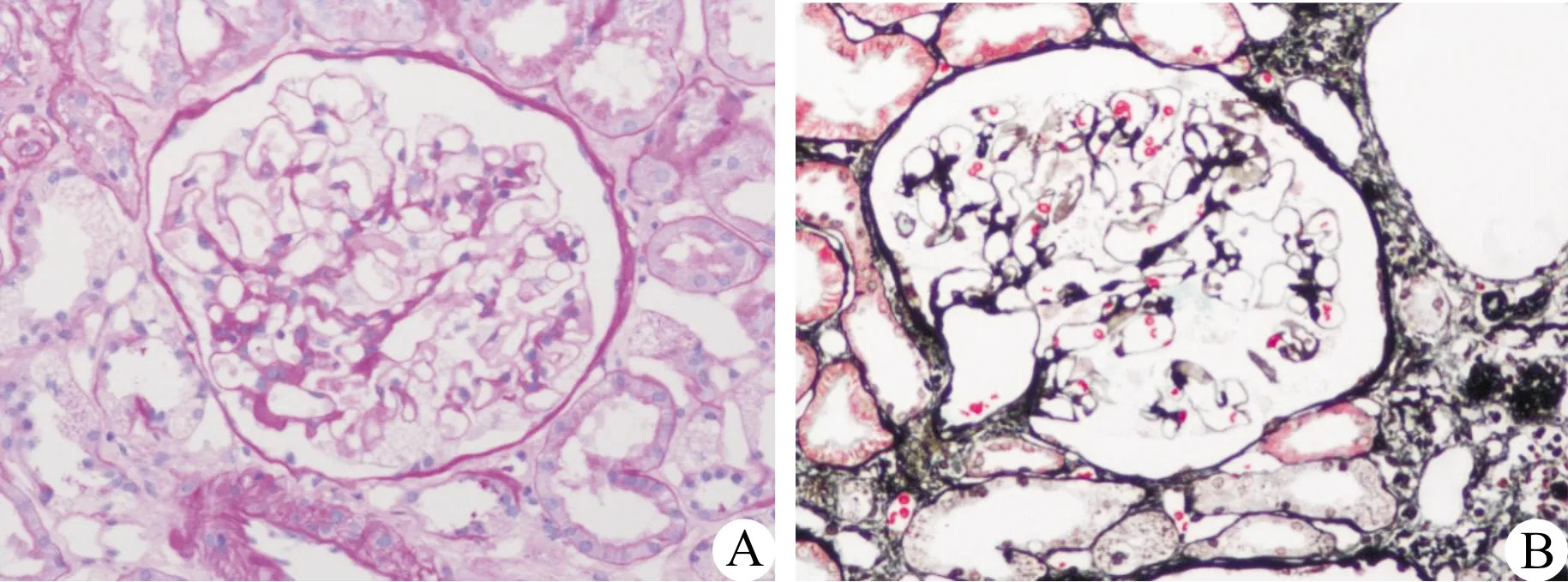
注:A:足细胞广泛空泡样变,部分肾小管上皮细胞空泡样变(PAS,×400);B:未见嗜复红物质沉积(PASM-Masson,×400)图1 Fabry病患者肾组织光镜病理图片
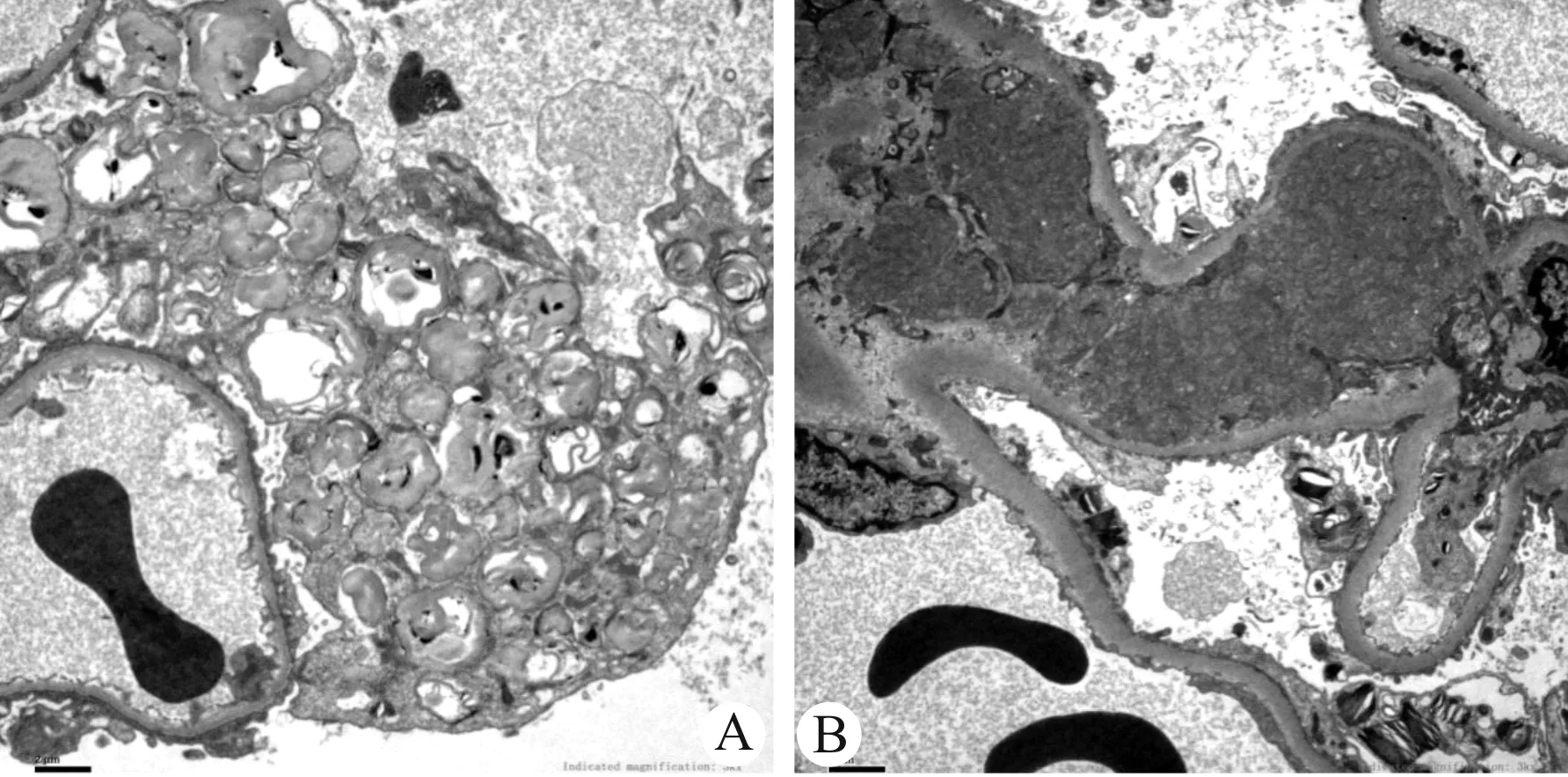
注:A:可见髓样小体(×3 000);B:可见斑马小体(×3 000)图2 Fabry病患者肾组织电镜病理图片
讨 论
Fabry病是一种X染色体连锁隐性遗传疾病,位于Xq22上的GLA基因发生突变,使得该基因编码的α-Gal A缺乏,鞘糖脂蓄积于肾脏、心脏、神经系统、皮肤、胃肠道、眼等组织细胞的溶酶体,从而导致细胞功能障碍,出现多系统受累。该病的患病率约1/117 000~1/40 000[1]。男性半合子起病往往早于女性杂合子,且病情重于女性。
由于代谢产物的蓄积是一个渐进的过程,因此临床表现也随着年龄的变化而会有所不同。Fabry病通常在儿童时期出现首发症状,表现为手足烧灼样疼痛或感觉异常,发热、应激、运动、饮酒均可诱发。其他症状还包括少汗或无汗,发热,冷/热不耐受,脂肪不耐受,餐后发作性腹痛、腹泻、恶心呕吐,皮肤血管角质瘤,角膜涡状混浊,耳鸣、听力下降等[2-3]。在Fabry病患者中多达50%男性和20%女性有肾脏受累的表现[4],光镜下可以看到组织细胞空泡样改变,电镜表现为典型的嗜锇性同心圆板层样包涵体(髓样小体和斑马小体),一般在20岁以后出现轻到中度蛋白尿,逐渐发展为肾衰竭。心脏和神经系统可出现左心室肥厚、心肌缺血、心律失常、心力衰竭、缺血性脑血管病等并发症[2-3]。对高危患者的筛查结果显示[2],Fabry病在年轻卒中患者中占0.5%,在高血压左心室肥厚中占0.9%,在特发性肥厚型心肌病中占0.5%~1%(男性为4%),在透析患者中占0.11%~0.17%。本例患者临床症状不明显,仅表现为轻中度蛋白尿,且否认家族史,追溯病史我们发现患者平素有手足灼热或感觉异常,心电图提示左心室高电压,这些非特异性的表现均与Fabry病有关,但均被忽视,若未行肾活检则会漏诊Fabry病。
基因检测是诊断Fabry病的金标准。轻度变异的患者通常有GLA基因错义突变和不同的酶活性残留[2]。酶活性低于25%~30%便可确诊,男性患者α-Gal A的活性常明显下降,而约30%的女性患者酶活性可以在正常范围[3]。如果是临床意义未明的GLA基因突变时,则在相关组织中发现三己糖苷酰基鞘鞍醇(Gb3或GL-3)水平升高将是最终的必要条件[2]。在鉴别诊断方面,需要注意排除由羟氯喹、氯喹和胺碘酮等药物诱导的磷脂沉积症[5]。我们对本例患者的家系(包括患者及其母亲、还有同母异父的姐姐和弟弟)进行了GLA基因检测,结果显示患者本人及其母亲、还有同母异父的弟弟均在同一个基因位点发现突变,除患者本人有临床表现外,母亲和弟弟的临床表型均为正常人群。从他们的基因图可以看到,GLA基因的EX6/CDS6存在错义突变,与互补DNA参考序列相比,第973位碱基的鸟嘌呤(guanine,G)被胞嘧啶(cytosine,C)取代,导致编码蛋白的第325位氨基酸由甘氨酸(glycine,Gly)转换成精氨酸(arginine,Arg)。由于GLA基因位于X染色体上,所以患者和弟弟均为半合子,母亲为携带有突变基因的杂合子,姐姐则不存在这一突变。该突变位点c.973G>C(p.Gly325Arg)在国际千人基因组计划数据库中的频率为0,目前在国内尚未报道,国外已有该位点在Fabry病患者中检出的文献报道[6],至于该突变位点是否为Fabry病的新发位点以及该位点的致病性还有待进一步研究。
Fabry病的特异性治疗首选酶替代疗法(enzyme replacement therapy,ERT)。临床试验和登记数据显示每两周一次使用α -半乳糖苷酶A(0.2 mg/kg)或β -半乳糖苷酶A(1.0 mg/kg)更有利于长期的肾脏转归,而且对于年轻患者获益最大[7]。一项观察性研究表明从足细胞中清除Gb3沉积物在年轻患者中是可以实现的,但是取决于累积的ERT剂量[8]。酶替代疗法需要终身每两周一次静脉输注,相比之下,酶增强治疗即隔日口服一次123 mg米加司他(migalastat)的方案更能被患者接受。在两项关键的3期临床研究中,无论是对于初治患者(FACETS研究:NCT00925301)或者对于从酶替代疗法中转换疗法的患者(ATTRACT研究:NCT01218659),米加司他均能有效地减少代谢底物并稳定肾功能[1,9]。其他非特异性治疗如血管紧张素转换酶抑制剂或血管紧张素II受体阻断剂,通过控制蛋白尿和高血压,对延缓Fabry病的肾功能进展也能起到一定的作用[7]。本例患者使用的是非特异性治疗,给予血管紧张素II受体阻断剂治疗后,蛋白尿有所减少。
总而言之,Fabry病患病率低,临床表现不典型,有些缺乏家族遗传史,临床上很容易被误诊和漏诊。对于不明原因的肾病患者应进行肾活检特别是电镜检查,若考虑为Fabry病则需进一步行α-Gal A酶活性测定和基因检测进行确诊。特异性治疗和非特异性治疗相结合才是治疗Fabry病的最佳方案。
猜你喜欢
分子催化(2022年1期)2022-11-02
上海交通大学学报(2021年8期)2021-09-02
烟草科技(2021年6期)2021-06-24
数字海洋与水下攻防(2021年2期)2021-05-08
哈尔滨工程大学学报(2021年1期)2021-02-25
水下无人系统学报(2019年1期)2019-03-15
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
中国医学创新(2018年28期)2018-02-23
云南中医中药杂志(2018年10期)2018-01-19
