IP-RP-HPLC 法同时测定感冒清片中3 种成分
2020-05-12 13:37朱链链王丽琼
中成药 2020年2期
朱链链,王丽琼
(1.乐山职业技术学院,四川 乐山 614000;2.乐山市食品药品检验检测中心,四川 乐山 614000)
感冒清片属于抗感冒药类中西药复方制剂,由3 种西药(对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏)和6 种中药(南板蓝根、大青叶、金盏银盘、岗梅、山芝麻、穿心莲叶)制成,该类制剂应用广泛,但其不良反应问题也居高不下[1],业界大多认为其原因是在临床使用时易忽略所含化学成分,关键项目(如有关物质、含有量均匀度、溶出度)缺失,故建议对该类药物进行再评价[2]。
文献[3-8]报道,采用HPLC 法测定对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏单一或多种成分含有量,但盐酸吗啉胍保留弱,易受溶剂峰干扰,而在流动相中加入离子对试剂可使分离变好。本实验建立离子对反相高效液相色谱(ion-pair reversephase high performance liquid chromatography,IP-RPHPLC)法同时测定感冒清片中对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏的含有量,并考察三者含有量均匀度,以期为该制剂质量标准提高提供依据。
1 材料
1.1 仪器 Agilent 1260 高效液相色谱仪(配置四元泵、DAD 检测器);XS205 电子天平、FE28 PH计(瑞士梅特勒-托利多公司);CQ-100B 超声波清洗机(上海跃进医用光学器械厂)。
1.2 试剂与药物 对乙酰氨基酚(100018-201610,质量分数99.9%)、盐酸吗啉胍(100483-200402,质量分数100%)、马来酸氯苯那敏(100047-201507,质量分数99.7%)对照品均购自中国食品药品检定研究院。感冒清片(广东一片天医药集团制药有限公司,批号171113、171208;广东南国药业有限公司,批号170606),每片含对乙酰氨基酚12 mg、盐酸吗啉胍12 mg、马来酸氯苯那敏0.5 mg。甲醇为色谱纯或分析纯;磷酸、磷酸二氢钾为分析纯;辛烷磺酸钠(离子对色谱试剂);水为纯化水。
2 方法与结果
2.1 色谱条件 Waters XTerraC18色谱柱(250 mm×4.6 mm,5 μm);流动相甲醇(A)-0.02 mol/L磷酸二氢钾(含0.005 mol/L 辛烷磺酸钠,10%磷酸调节pH 至3.0)(B),梯度洗脱(0~20 min,10%~75% A;20~21 min,75%~10% A;21~26 min,10% A);体积流量1.0 mL/min;柱温30 ℃;检测波长215 nm;进样量10 μL。色谱图见图1。由此可知,各成分分离度良好,理论塔板数按对乙酰氨基酚峰计为18 394,其他成分在三者保留时间上没有色谱峰,不干扰测定。
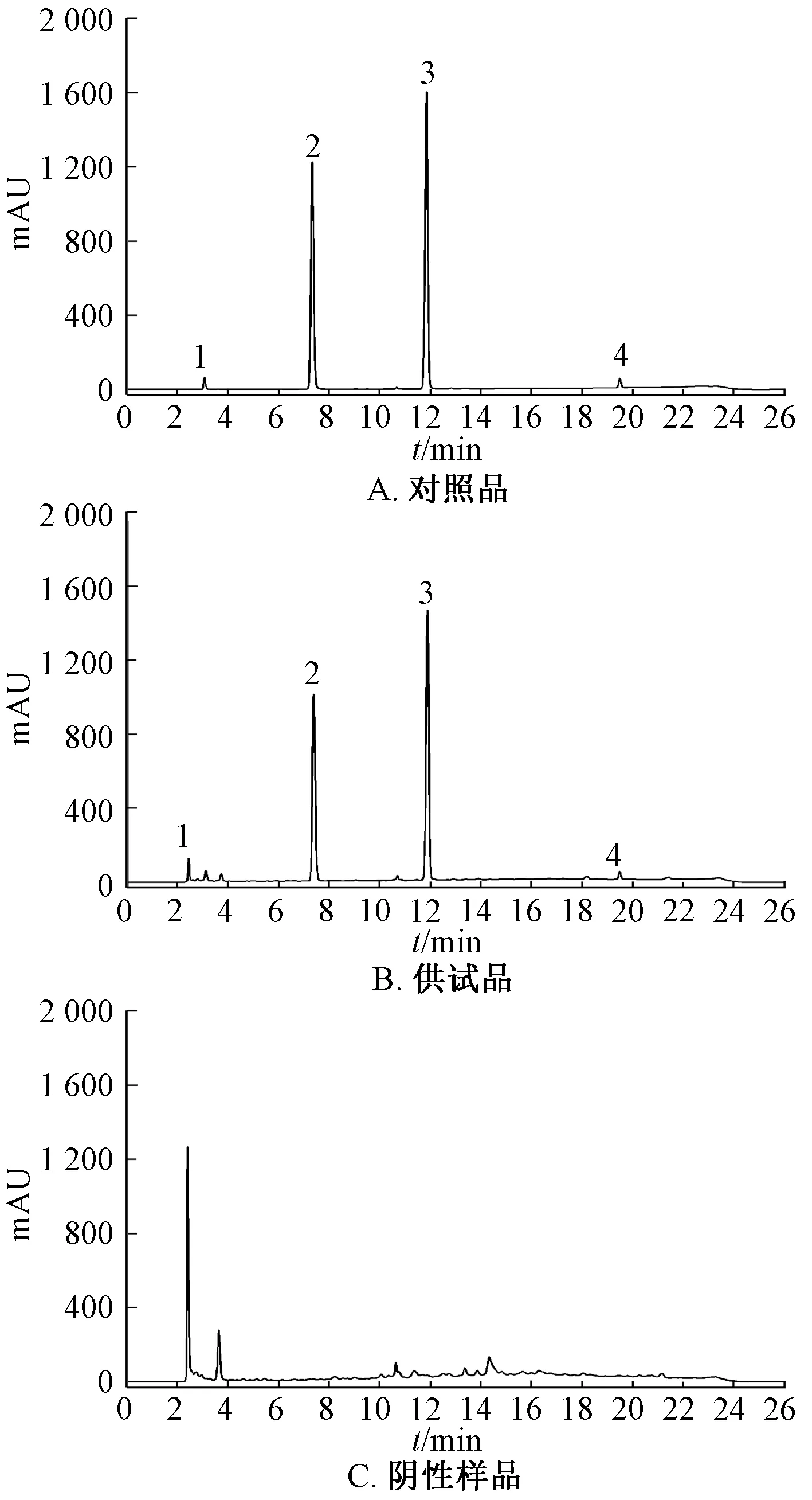
图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.2 溶液制备
2.2.1 对照品溶液 精密称取对照品对乙酰氨基酚48.38 mg、盐酸吗啉胍47.74 mg,置于同一50 mL量瓶中,25%甲醇(pH=3.0)溶解;精密称取马来酸氯苯那敏对照品10.24 mg,置于10 mL 量瓶中,25%甲醇(pH=3)溶解并稀释至刻度,摇匀,精密量取2 mL,置于上述50 mL 量瓶中,溶剂稀释至刻度,作为贮备液。精密量取5 mL 于10 mL 量瓶中,25%甲醇(pH=3)稀释至刻度,摇匀,即得。
2.2.2 供试品溶液 取本品10 片,精密称定质量,计算平均片重,研细,取约0.22 g(相当于12 mg 对乙酰氨基酚),置于25 mL 量瓶中,25%甲醇(pH=3)超声助溶,放冷,25%甲醇(pH=3)稀释至刻度,摇匀,滤过,即得。
2.2.3 阴性样品溶液 按处方比例分别制备不含对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏的阴性样品,按“2.2.2”项下方法制备,即得。
2.3 定量限 精密称取对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏对照品适量,25%甲醇(pH=3)溶解并分别稀释成0.32、0.05、0.51 μg/mL,在“2.1”项色谱条件下进样测定。结果,三者信噪比(S/N)分别为37.2、43.8、46.8(以氯苯那敏峰计);以S/N=10,得定量限分别为0.09、0.01、0.11 μg/mL。
2.4 线性关系考察 精密量取对照品溶液1、2、4、6、8 mL,置于10 mL 量瓶中,25%甲醇(pH=3)稀释至刻度,摇匀,得线标溶液,取其与对照品溶液共6 份,在“2.1”项色谱条件下进样测定。以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,结果见表1,可知各成分在各自范围内线性关系良好。
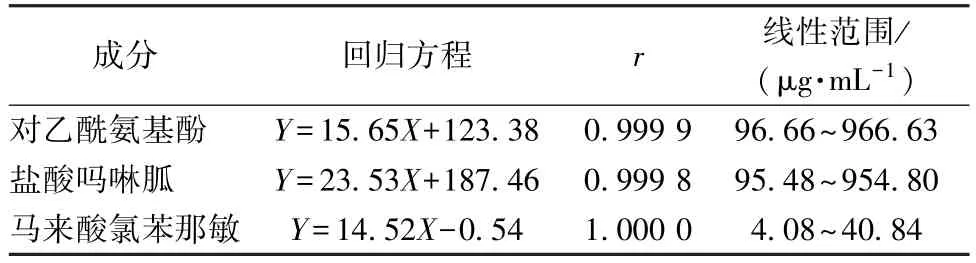
表1 各成分线性关系Tab.1 Linear relationships of various constituents
2.5 精密度试验 精密量取“2.2.2”项下对照品溶液,在“2.1”项色谱条件下进样测定6 次,测得对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏峰面积RSD 分别为0.2%、0.2%、0.4%,表明仪器精密度良好。
2.6 重复性试验 取同一批本品(批号171113),按“2.2.2”项下方法平行制备6 份供试品溶液,在“2.1”项色谱条件下进样测定,测得对乙酰氨基酚、盐酸吗啉胍、马来酸氯苯那敏(以氯苯那敏计)含有量RSD 分别为1.2%,1.0%、1.3%,表明该方法重复性良好。
2.7 稳定性试验 精密量取“2.2.2”项下供试品溶液,室温下于0、2、4、6、8、12、24 h 在“2.1”项色谱条件下进样测定,测得对乙酰氨基酚、盐酸吗啉胍、氯苯那敏峰面积RSD 分别为0.4%,0.6%、1.1%,表明供试品溶液在24 h 内稳定性良好。
2.8 加样回收率试验 精密称取含有量已知的本品6 份(批号171113),每份约0.11 g,置于6 个25 mL 量瓶中;精密称取对照品对乙酰氨基酚60.21 mg、盐酸吗啉胍60.02 mg,置同一10 mL 量瓶中,25%甲醇(pH=3)溶解并稀释至刻度,精密量取1 mL,置于上述25 mL 量瓶中;精密称取马来酸氯苯那敏对照品12.51 mg,置于50 mL 量瓶中,25%甲醇(pH=3)溶解并稀释至刻度,精密量 取1 mL,置于上 述25 mL 量瓶中,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,计算回收率,结果见表2。
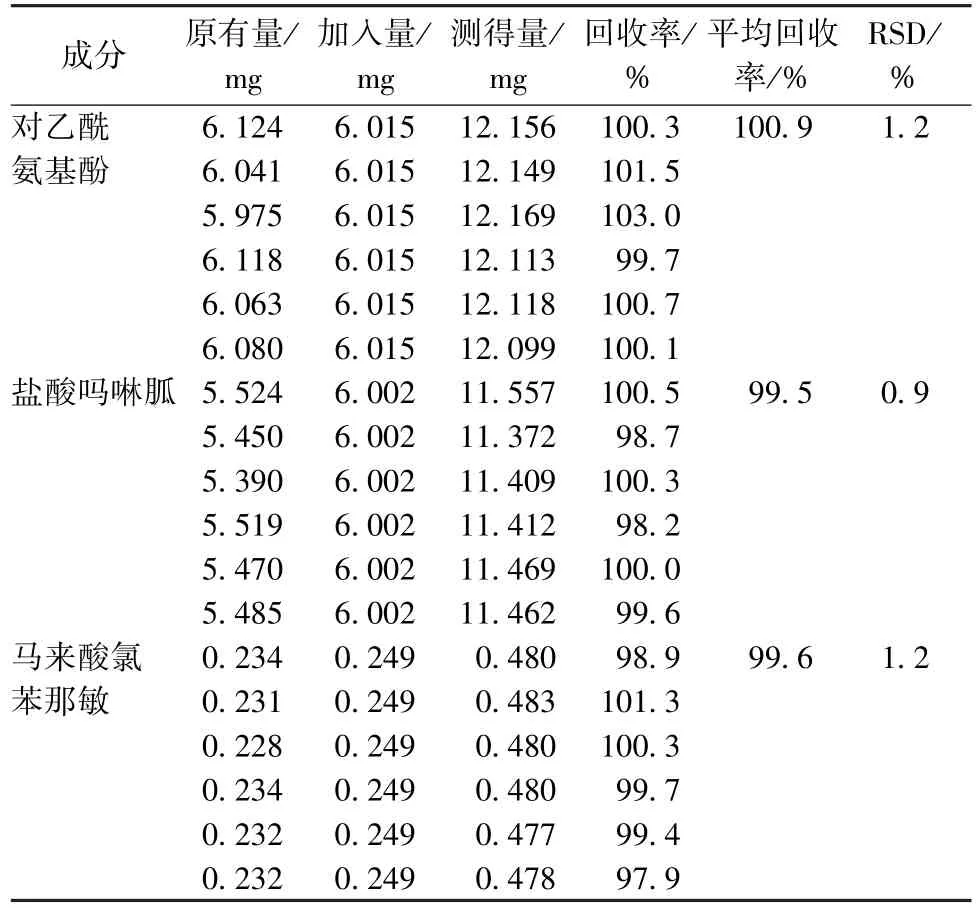
表2 各成分加样回收率试验结果(n=6)Tab.2 Results of recovery tests for various constituents(n=6)
2.9 样品含有量测定 精密称取本品3 批,每批3 份,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,外标法计算含有量,结果见表3。

表3 各成分含有量测定结果(n=3)Tab.3 Results of content determination of various constituents(n=3)
2.10 含有量均匀度测定 取本品3 批,每批10份,置于25 mL 量瓶中,25%甲醇(pH=3)超声助溶,放冷,25%甲醇(pH=3)稀释至刻度,摇匀,各精密量取10 μL,在“2.1”项色谱条件下进样测定,外标法计算含有量。然后,根据2015年版《中国药典》 四部方法[9],计算A[标示量(100)与含有量平均值之差的绝对值]+2.2S(含有量标准差),结果见表4,发现均符合药典规定[A+2.2S<L(L以15.0 计)]。
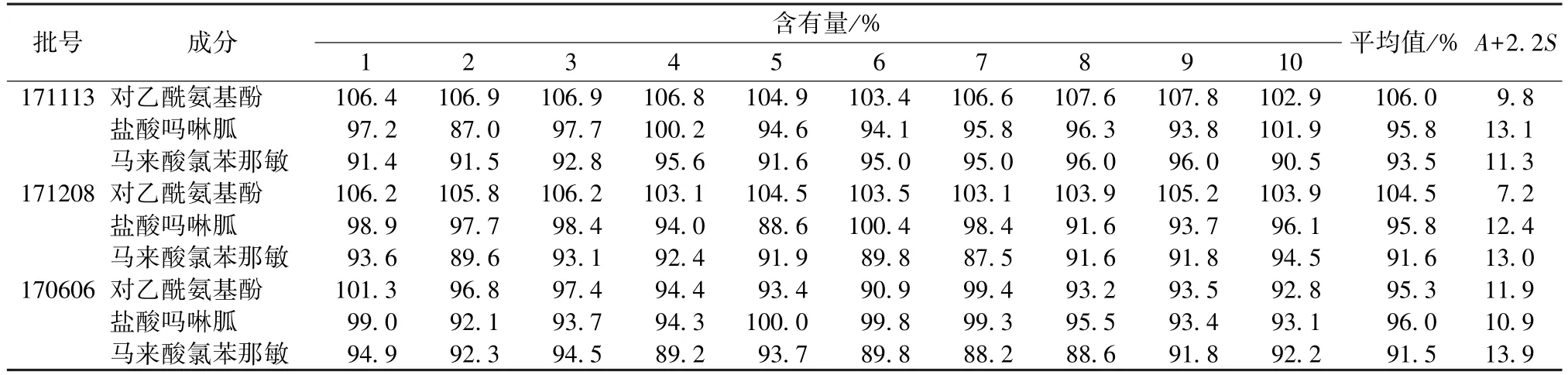
表4 各成分含有量均匀度测定结果Tab.4 Results of content uniformity determination of various constituents
3 讨论
3.1 检测波长选择 马来酸氯苯那敏最大吸收波长为262 nm,但供试品溶液中其质量浓度较低,故考虑215 nm,同时在该处对乙酰氨基酚有波谷,盐酸吗啉胍有波峰。最终,确定215 nm 作为检测波长[10]。
3.2 流动相选择
3.2.1 离子对试剂种类与浓度 本实验考察了戊烷磺酸钠、己烷磺酸钠、庚烷磺酸钠、辛烷磺酸钠,发现离子对试剂烷基链越长,盐酸吗啉胍保留时间越长,与相邻的对乙酰氨基酚色谱峰分离度越高[11-12],而且其浓度对色谱峰保留也有很大影响[13],故又对此进行考察。结果显示,随着离子对试剂浓度的升高,盐酸吗啉胍峰保留时间延长,峰型由不对称逐渐优化为对称,当辛烷磺酸钠浓度高于0.005 mol/L 时,分离度合适,峰形良好。同时,考虑到离子对试剂浓度过大时对仪器和色谱柱均有不利影响,故最终选择0.005 mol/L 辛烷磺酸钠,可使盐酸吗啉胍在色谱柱上有较好的保留,并与其他成分的分离度理想,互不干扰。
3.2.2 缓冲液 本实验考察了缓冲盐对成分分析的影响,发现基线在无磷酸盐的流动相中不平稳,峰形不对称;添加磷酸二氢钾后基线平稳,而且由于离子强度的改变,盐酸吗啉胍色谱峰保留时间提前,故选择0.02 mol/L 磷酸二氢钾作为缓冲液。
3.2.3 流动相pH 本实验考察了2.0、3.0、4.0、4.5,发现随着pH 降低盐酸吗啉胍保留时间延长,同时峰形高而尖锐,考虑到C18色谱柱对pH 值的耐受范围为2~8,故最终选择3.0 作为流动相pH。
3.3 柱温选择 柱温对离子对色谱的影响较大[14]。本实验考察了20、25、30、35、40 ℃,发现柱温越高,各组分出峰越快,但过高时氯苯那敏色谱峰受干扰较大,峰形较差。最终,选择30 ℃作为柱温,此时出峰时间适中,峰形理想。
猜你喜欢
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
药品评价(2021年17期)2021-11-06
化学工程师(2021年9期)2021-10-14
环境卫生工程(2021年1期)2021-03-19
世界农药(2020年12期)2021-01-04
分析仪器(2020年5期)2020-11-09
土壤(2019年6期)2020-01-06
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年12期)2019-05-20