
基于CRISPR/Cas9 技术快速构建PRV gE 基因缺失病毒
2020-05-09 01:04
中国动物检疫 2020年5期
(内蒙古农业大学兽医学院,农业农村部动物疾病临床诊疗技术重点实验室,内蒙古呼和浩特 010018)
伪狂犬病病毒(pseudorabies virus,PRV)属于疱疹病毒科,会导致成年母猪流产、产死胎以及仔猪中枢神经系统疾病,未免疫母猪产下的仔猪感染PRV 时,死亡率可达100%[1-2]。早在1947 年,我国首次从患猫体内检测出PRV。1970 年后,PRV 在我国呈蔓延趋势,直至从匈牙利引入Bartha-K61 株疫苗后,疫情才得到有效控制[3]。2011 年以来,PRV 在我国发生变异,使Bartha-K61 疫苗不能提供足够的保护,导致伪狂犬病迅速流行,并席卷全国[4-7],其中母猪患病率达35%以上,野毒感染猪群超过50%,对我国养猪业造成了巨大损失[8]。PRV 基因组编码许多蛋白,其中gE 蛋白是主要的毒力因子。在PRV 变异株感染的猪群中,检测发现gE基因抗体水平逐年上升。因此,gE基因缺失疫苗可以在降低病毒毒力的同时区分野毒感染和疫苗免疫[9]。我国最初引入的Bartha-K61 株就是gE基因缺失疫苗株。早在2000年,何启盖等[10]使用TK-/gG-/LacZ+突变株进行动物实验,证实PRV 基因缺失毒株的安全范围比亲本毒株广,具有很好的免疫原性。自2013 年以来,CRISPR/Cas9 技术作为强大的基因编辑技术[11],已经在病毒基因编辑上获得了广泛应用,如:Van等[12]利用CRISPR/Cas9 技术成功编辑了HCMV和HSV-1;2018 年穆艳霞等[13]使用CRISPR/Cas9技术,利用先重组EGFP 标签反向筛选的方法成功获取了IBR-ΔgE病毒;汤艳东等[14]构建了萤光素酶标记基因、EGFP 基因双标签重组病毒,同时建立了PRV 大片段缺失平台,证实了2 个sgRNA介导的大片段基因缺失效率更高。因此,本研究基于CRISPR/Cas9 技术与实验室分离到的PRV 毒株,快速对其gE基因进行编辑,旨在为今后的PRV 基因编辑提供思路,也为后期应对PRV 变异株的基础研究及防控提供一种候选方法。
1 材料与方法
1.1 材料
1.1.1 病毒与细胞系 PRV-1 毒株,由内蒙古农业大学兽医学院传染病教研室分离、鉴定及保存;PK-15(猪肾细胞)、VERO(非洲绿猴肾细胞),由内蒙古农业大学传染病教研室保存。
1.1.2 主要试剂及载体 胎牛血清(fetal bovine serum,FBS),DMEM 培养基,opti-MEM 以及EDTA-胰酶,购自Gibco 公司;BbsI 限制性核酸内切酶,购自NEB 公司;PrimeSTAR GXL DNA Polymerase 高保真酶、solution I,购自TaKaRa 公司;DNA 提取试剂盒、胶回收试剂盒,购自Therom公司;病毒基因组DNA/RNA 提取试剂盒,购自TIANGEN 公司;低熔点琼脂糖,购自Sigma 公司;转染试剂Lipofactamine 3 000,购自Thermo Fisher公司;大肠杆菌DH5ɑ 感受态细胞、Stbl 3 化学感受态细胞,购自北京全式金生物技术有限公司;pcDNA3.1载体、pSpCas9-2A-puro 和pSpCas9(BB)-2A-GFP 载体,由内蒙古农业大学传染病教研室保存。
1.2 方法
1.2.1 PRVgE基因克隆测序 根据NCBI 上已公布的伪狂犬病毒(PRV)基因序列(登录号KU056477.1),在primer premier 5.0 软件上设计针对gE基因的上下游引物PRV-gE-F、PRV-gE-R(表1);以本实验室提取的PRV-1 基因组为模板,采用PCR 方法扩增gE基因。PCR 反应体系:ddH2O 15 μL、GXL buffer 5 μL、dNTP 2 μL 以及PRV-gE-F、PRV-gE-R 各1 μL,PRV-1 基因组0.75 μL,GXL DNA Polymerase 酶0.25 μL。PCR反应条件:94 ℃预变性120 s;98 ℃变性10 s,60 ℃退火15 s,68 ℃延伸120 s,35 个循环;68 ℃终延伸10 min,4 ℃保存。PCR 产物送华大基因有限公司测序。
1.2.2 野生病毒感染力测定 将PRV-1 病毒按照10-1~10-8进行10 倍倍比稀释后,各吸取100 μL 病毒稀释液,接种于长到80%左右的PK-15 细胞上,每组8 个重复,同时设置阴性对照。逐日观察并记录结果,5 d 后按照Reed-Muench 法进行TCID50测定。计算公式为:
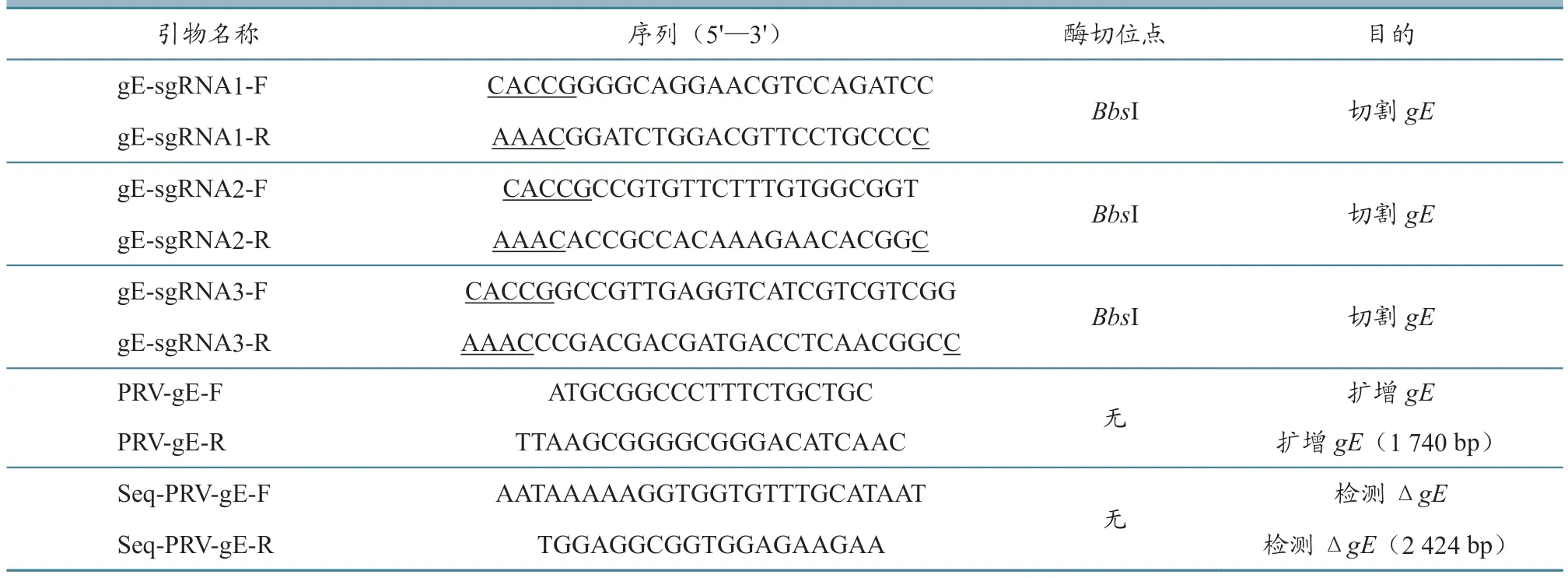
表1 试验所用引物及目的

将比距加在高于50%死亡稀释度的对数上,即为0.1 mL 所含病毒液的稀释度。
1.2.3 转染细胞系筛选 首先将pSpCas9(BB)-2A-GFP 载体,利用脂质体转染法(Lipofectamine®3 000),将PX459-1、PX459-2 和PX459-3 质粒分别转染PK-15 细胞与VERO 细胞,48 h 后荧光显微镜(ZEISS、HAL100)下观察,筛选转染效率较高的细胞系。
1.2.4 CRISPR/Cas9 载体构建及筛选 按照1.2.1测序得到PRVgE基因序列,通过网站http://crispr.mit.edu/设计3 对分数较高且GC 含量约50%的gE-sgRNA,并合成其互补引物对(表1);将互补的gE-sgRNA 引物gE-sgRNA1-F/gE-sgRNA1-R、gE-sgRNA2-F/gE-sgRNA2-R 和gE-sgRNA3-F/gEsgRNA3-R 进行95 ℃ 5 min 高温变性,然后逐渐降温至16 ℃,获得3 条双链sgRNA;以BbsI 双酶切pSpCas9-2A-puro 载体,胶回收大的载体片段;将3 条双链gE-sgRNA 连入pSpCas9-2A-puro 酶切回收的载体中,使用Solution I 16 ℃过夜连接,将连接产物转化Stbl 3 感受态细胞,提取质粒,对测序结果使用snapgene 软件分析,获得CRISPR/Cas9 重组质粒PX459-1、PX459-2 和PX459-3。将构建好的PX459-1、PX459-2 和PX459-3 分别转染VERO 细胞,12 h 后接种MOI=0.1 的PRV-1,感染48 h 后使用10%中性甲醛固定2 h,之后使用0.8%结晶紫染色0.5 h,冲洗干净后观察。
1.2.5 PRV-1-ΔgE构建 利用脂质体转染法,将CRISPR/Cas9 载体PX459-1、PX459-2,按 照1:1 的比例转染入VERO 细胞培养6 h,更换含2%FBS 的细胞维持液,培养至12 h 后接种MOI=0.1的PRV-1,待细胞病变至90%左右时收毒;将病毒反复冻融3 次,按照1:1 000 稀释度稀释感染6 孔板中的PK-15 细胞,病毒吸附2 h 后,使用DMEM(含2% FBS、1%低熔点琼脂糖)覆盖,置于37 ℃、5%的CO2培养箱中培养48 h;于超净工作台中使用枪头随机挑起10 个病毒蚀斑,分别置于200 μL DMEM 中,反复冻融3 次后接种生长至90%的PK-15 细胞。重复以上步骤,噬斑克隆纯化5 代,然后将病毒接种于PK-15 细胞中扩大培养,提取病毒基因组,使用检测引物seq-PRVgE-F/R 进行PCR 扩增检测。PCR 体系与反应条件与1.2.1 中介绍相同。将测序结果使用MEGA 7 软件进行对比分析,鉴定后扩大培养,-80 ℃保存。
1.2.6 PRV-1-ΔgE遗传稳定性检测 将PRV-1-ΔgE接种至PK-15 细胞,连续传代至第20 代,并取第5、10、20 代缺失病毒DNA,使用PRV-gE-F和PRV-gE-R 进行PCR 扩增。PCR 产物送华大基因测序,并对测序结果进行比对。
2 结果与分析
2.1 PRV-1 gE 基因PCR 扩增
按照1.2.1 中PCR 体系获得的gE基因测序结果为1 740 bp,与试验预期一致(图1)。
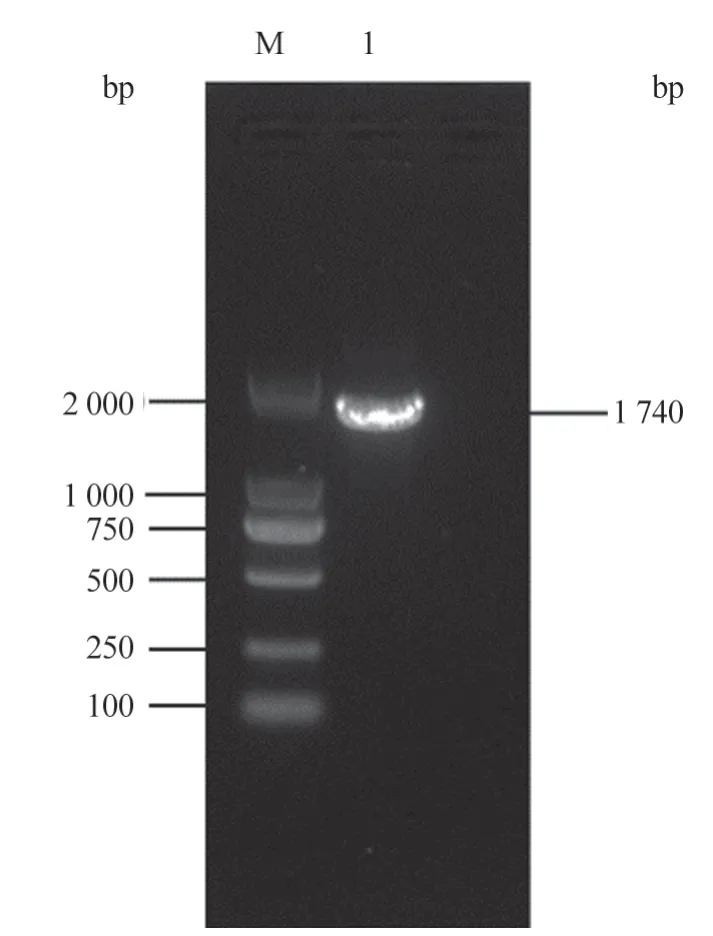
图1 PRV gE 基因PCR 扩增结果
2.2 PRV 野毒感染力测定
野生病毒感染力测定如表2 所示。当病毒稀释至10-6时,出现CPE 的细胞孔所占百分比高于50%,为76.9%,稀释至10-7时,出现CPE 的细胞孔所占百分比低于50%,为36.4%。
将表2 所测得数据带入1.2.2 中的公式,得到的比距约为0.7,加上高于50%感染的稀释度对数(10-6),PRV-1 的TCID50为10-7.7/mL。
2.3 转染细胞系筛选
通过荧光显微镜观察,发现VERO 细胞转染效率高于PK-15 细胞(图2),因此本试验选择VERO 细胞作为转染细胞系。
2.4 CRISPR/Cas9 质粒构建与筛选
根据snapgene 软件分析结果,判定CRISPR/Cas9 质粒PX459-1、X459-2 和PX459-3 构建成功(图3)。将PX459-1、X459-2 和PX459-3 质 粒转染细胞筛选,发现sgRNA1 和sgRNA2 效率大于sgRNA3(图4),因此选择sgRNA1 和sgRNA2做后续试验。
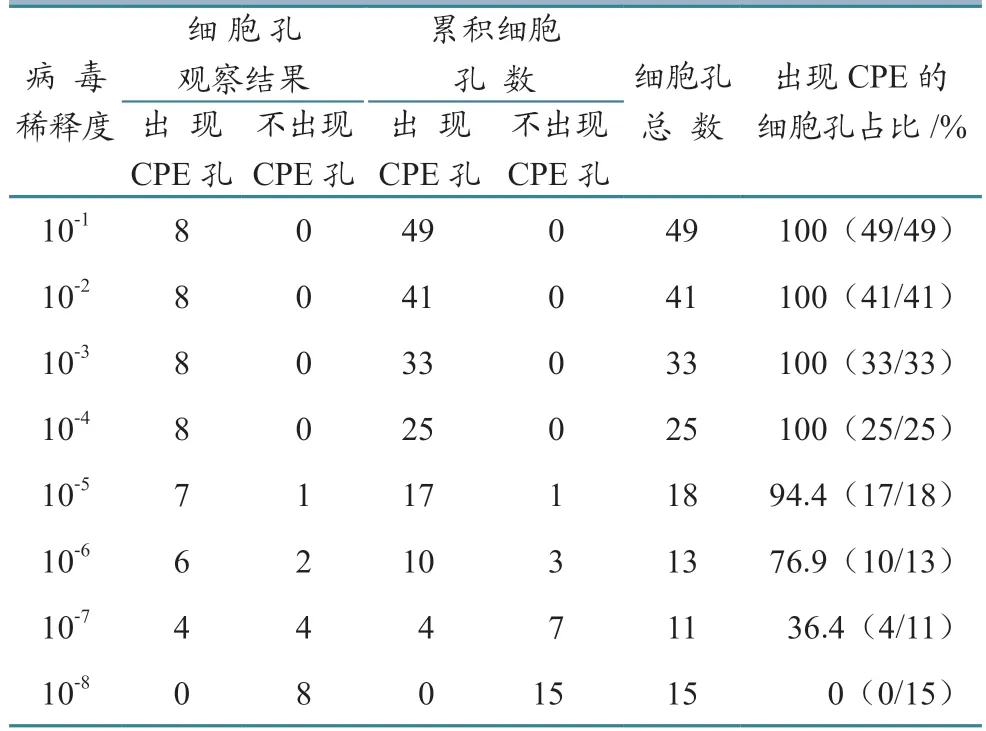
表2 病毒TCID50 测定结果
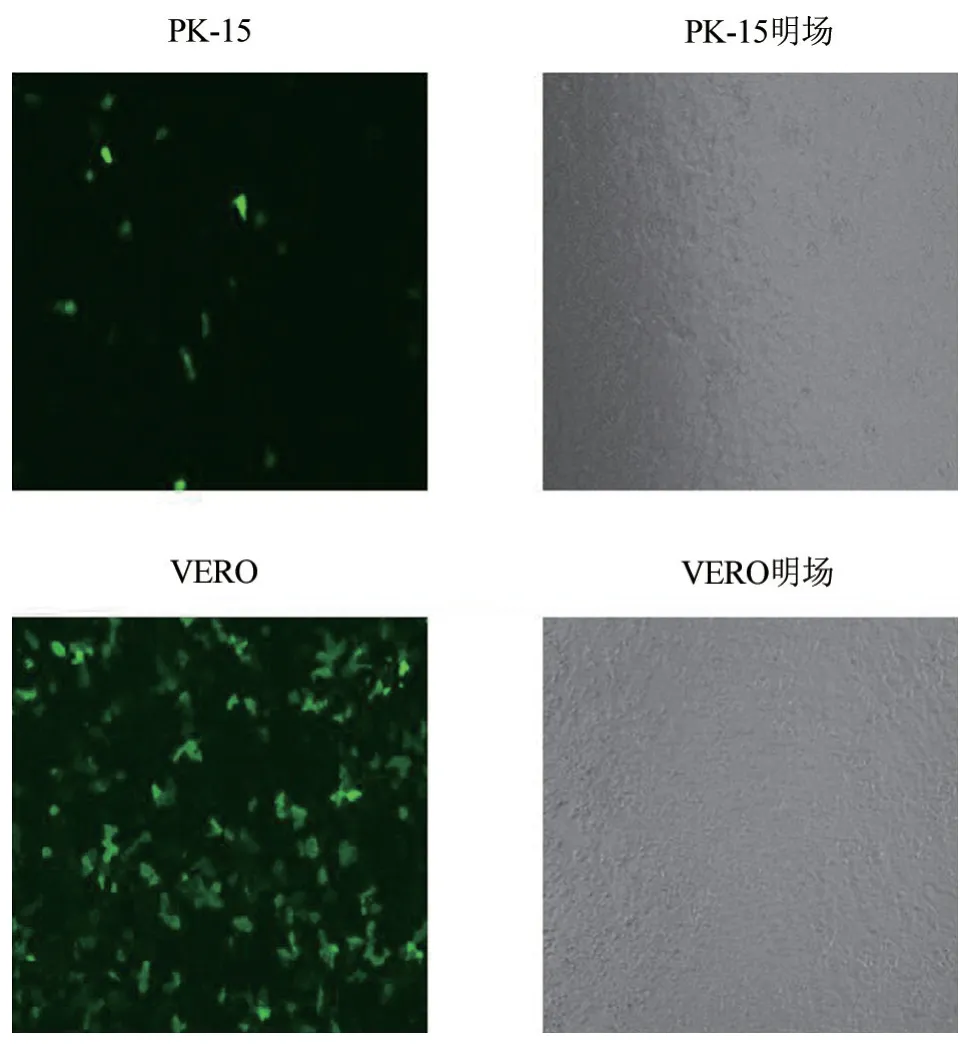
图2 pSpCas9(BB)-2A-GFP 在PK-15 与VERO 细胞上的转染效率(50×)
2.5 PRV-1-ΔgE 构建
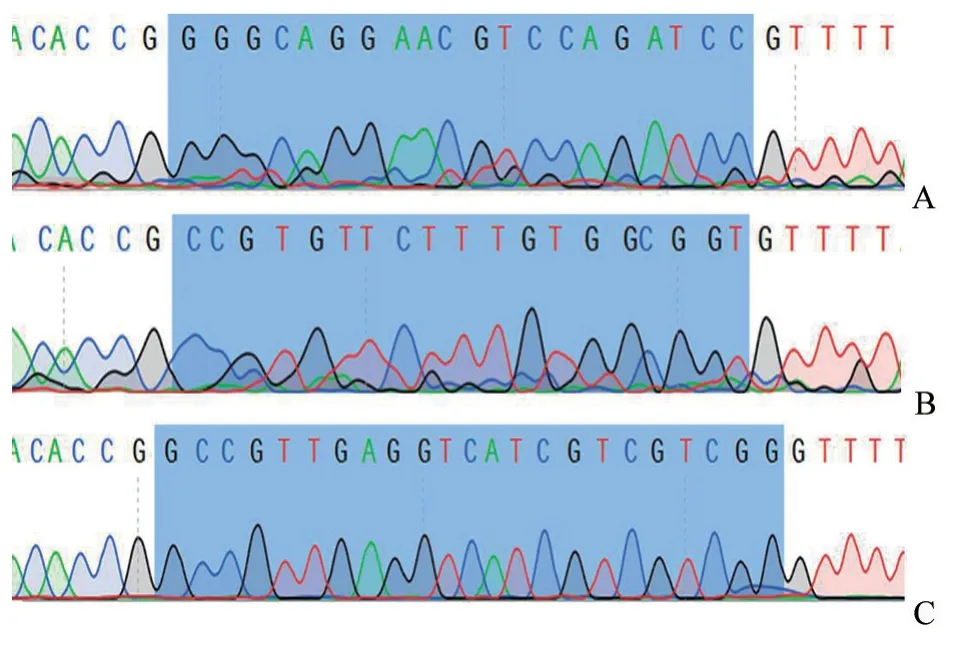
图3 sgRNA 构建测序结果
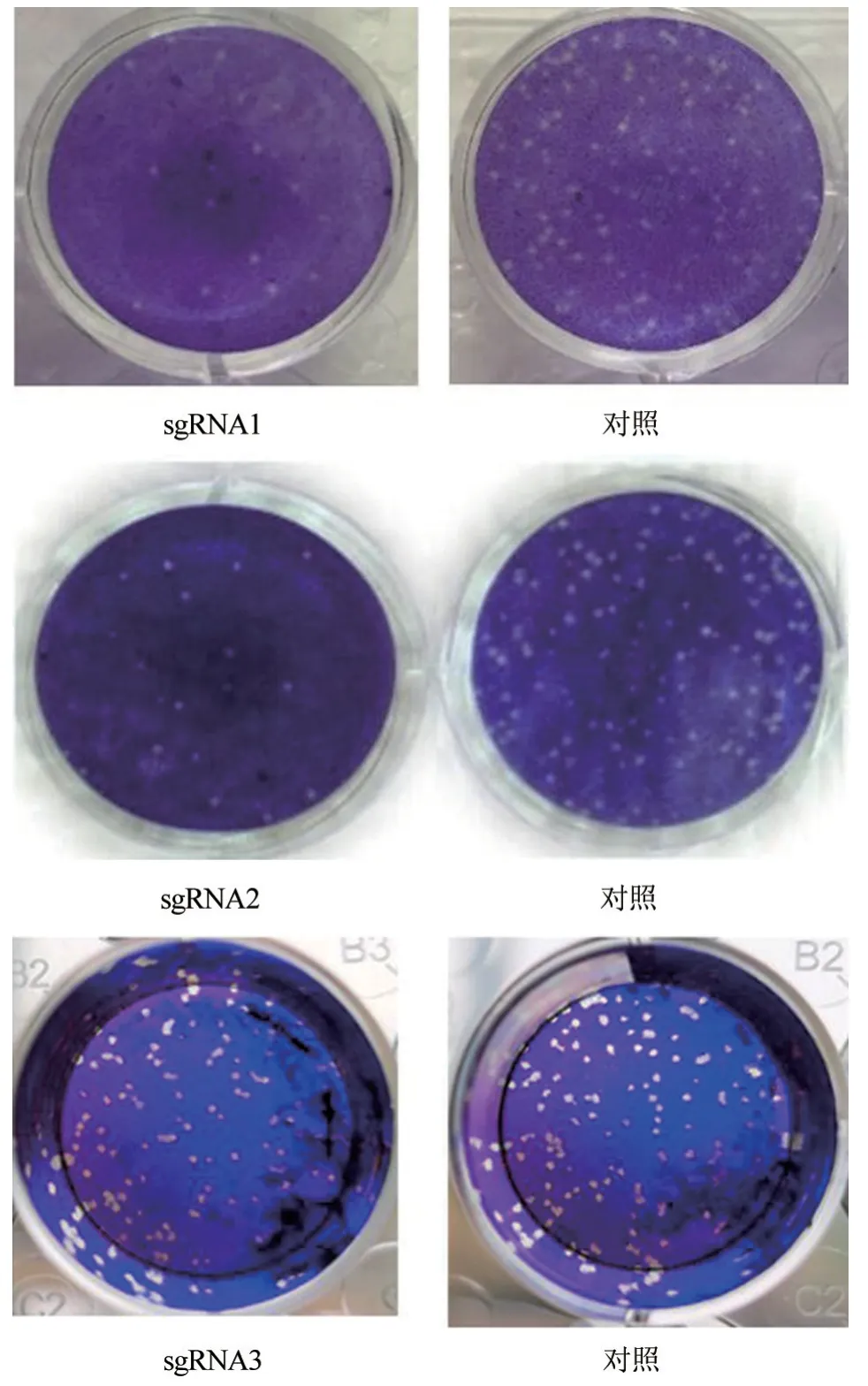
图4 sgRNA 筛选结果
使用seq-PRV-gE-F/R 引物进行PCR 检测,发现gE基因发生缺失(图5);将测序结果使用MEGA 7 软件比对分析发现,PCR 产物大小为2 424 bp,与预期一致,PRV-1-ΔgE为2 132 bp,gE基因323~613 bp 位置缺失291 bp(图6)。
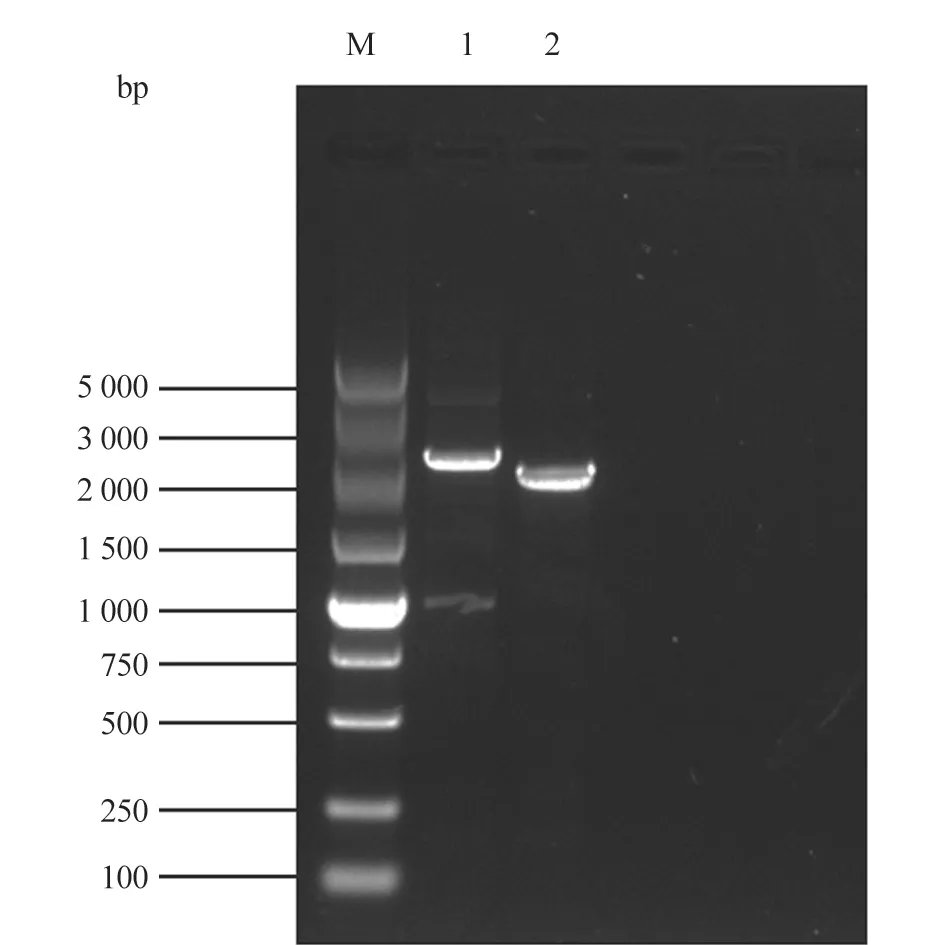
图5 PRV gE 基因检测引物PCR 鉴定结果
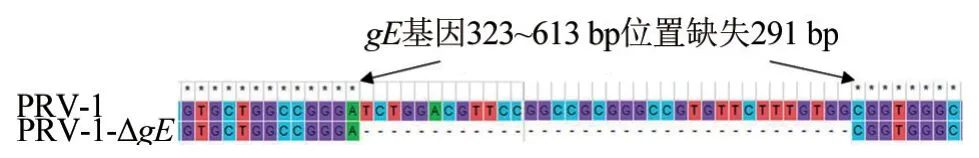
图6 PRV-1 与PRV-1-ΔgE 测序比对结果
2.6 PRV-1-ΔgE 遗传稳定性检测
将PRV-1-ΔgE连续传代至20 代,分别取5、10、20 代病毒进行测序分析,发现没有发生碱基突变现象。
3 讨论
目前,针对PRV 感染还没有系统有效的治疗方法,因此疫苗接种是控制其流行和净化的有效方法。自2011 年以来,PRV 不断发生变异,至今依然在我国部分地区广泛流行[15-16],因此快速应对新发变异的PRV 就显得尤为重要。这些基因的缺失会导致PRV 毒力降低,且不影响病毒的感染和复制能力。据相关资料[17-18]显示,一些欧美国家通过基因缺失疫苗免疫,已经根除了PRV。PRV 基因工程疫苗研制通常选用缺失TK、gE、gI或gG等毒力基因[19-20]。
穆艳霞等[13]使用CRISPR/Cas9 技术编辑IBRV,在gE基因位置处重组EGFP 荧光标记,通过sgRNA 反向敲除EGFP 基因构建IBR-ΔgE病毒。吴凤笋[21]使用传统的同源重组结合Cre/loxp系统,先在gE、TK位置分别重组EGFP 标记基因,筛选携带荧光的PRV,后将标记基因分别切除构建了1 株PRV-ΔgE-ΔTK重组病毒。靶向基因编辑技术与传统的同源重组技术相比较,它对基因组的修饰效率可以提高103~105倍[22]。因此,本研究基于CRISPR/Cas9 技术,仅通过单独转染1 对sgRNA,接种病毒诱导细胞内非同源末端修复即获得PRV-1-ΔgE。相比较于传统的同源重组技术,CRISPR/Cas9 技术大大提高了基因编辑筛选速度与效率。首先,在CRISPR/Cas9 技术当中,筛选转染效率高的细胞系与获得高效的sgRNA至关重要。因此,本试验首先选择了PK-15 细胞与VREO 细胞作为转染细胞系,经过筛选发现VERO 细胞的转染效率远高于PK-15 细胞且可以被PRV 感染。本试验结果与汤艳东[13]、穆艳霞[14]等细胞系筛选结果一致。其次,sgRNA 的工作效率及脱靶效率直接影响基因编辑的成功率。Cho 等[23]认为成对的Cas9 内切酶共同作用于目标基因组,会使脱靶效率明显降低。在sgRNA 筛选中,本试验首先设计了3 条sgRNA,经过噬斑形成试验,筛选到2条可以高效切割病毒基因的sgRNA,最后在噬斑克隆过程中,因为没有标签,所以随机筛选了10个单独噬斑克隆。在这个过程中,要挑选相对于野生病毒噬斑稍小的噬斑克隆,突变毒株的噬斑会稍小于野生病毒[14]。同时,相比较于标签反向筛选,本试验没有插入标签,因而后期就省去了删除标签的步骤。
4 结论
综上,本试验采用CRISPR/Cas9 第3 代基因编辑技术,快速对PRV-1 进行编辑,确定了1对高效编辑PRVgE基因的sgRNA,并获得1 株PRV-1-ΔgE。本研究提供了一种高效的PRV-1 毒株编辑方法,为后续构建多基因缺失病毒提供了基础数据,也为快速应对PRV 变异及其相关变异株基础研究提供了新思路。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
军事文摘(2022年16期)2022-08-24
科学与生活(2021年16期)2021-11-25
中国种业(2021年11期)2021-11-25
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
浙江医学(2020年19期)2020-10-20
生物学教学(2019年3期)2019-03-22
