
多吡啶铁配合物制备及电催化质子还原性能
2020-05-08 02:48荣冰莹宋丽娟
石油化工高等学校学报 2020年2期
荣冰莹,刘 贺,惠 宇,宋丽娟,钟 伟
(1.辽宁石油化工大学辽宁省石油化工催化科学与技术重点实验室,辽宁抚顺113001;2.嘉兴学院生物与化学工程学院,浙江嘉兴314001)
众所周知,能源危机已成为制约人类发展和进步的关键问题。人类依赖的化石能源(煤、石油、天然气)不仅资源有限,而且燃烧时产生大量的温室气体CO2和其他有害物质[1]。探索和研究可持续、环境友好的新型能源以减轻乃至完全脱离对化石能源的依赖已成为各国科技工作者必须面对的挑战[2-3]。作为一种清洁无污染的新能源,氢能源具有无可比拟的潜在开发价值,而廉价高效的制氢催化剂则是氢能开发与利用的关键。
目前,工业生产中最有效的制氢催化剂是铂、钯等贵金属,但这些金属在地壳中的丰度极低,价格昂贵,无法满足可持续发展的要求。另一方面,自然界的藻类和微生物中存在一类金属酶——氢化酶,可以高效、快速和可逆地催化质子还原制氢和氢气氧化反应[4],为开发廉价高效的制氢催化剂提供了可借鉴的思路。事实上,受氢化酶的启发,化学工作者已经开始尝试用化学方法探索合成低成本和高效率的制氢催化剂[4]。自从20世纪末[FeFe]-氢化酶的详细晶体结构被解析以来[5-6],国内外化学工作者合成了许多二铁羰基化合物,作为该酶金属中心的模型化合物并开展了相关性质研究[7-8]。C.Nagoshi等[4]在[FeFe]-氢化酶的仿生模拟及质子催化电荷转移机理方面开展了系统深入的研究。
迄今为止,尽管研究者在[FeFe]-氢化酶仿生制氢催化体系方面开展了卓有成效的研究,但已报道的这些催化体系大多合成步骤繁琐或条件苛刻,催化效果难与自然界[FeFe]-氢化酶相媲美[9-10]。因此,许多研究小组着眼于开发新型廉价、简单方便的制氢催化体系,以期达到在低电位条件下高效催化制氢的目的。最近几年以来,科研人员利用地球上比较丰富且廉价易得的金属离子如钴[11]、镍[12]、铜[13]、铁[14-16]等,设计合成新型的电催化分子催化剂,在催化还原质子制氢方面取得了明显的成效。如R.Tatematsu等[12]报道了一种含有机膦配位的Ni(II)化合物,其配体具有稳定镍的氧化态且作为质子转移位点的碱的作用,在冰醋酸介质中,可以在较低的过电位下(590 mV)高效地催化制氢,TOF达到8 400 s-1。W.R.McNamara课题组报道的铁-多吡啶基配合物在乙腈溶液电催化质子还原制氢[16],TOF达1 000 s-1,过电位为660 mV;在加入水作为质子源后,TOF可达3 000 s-1,成为目前报道的活性较高的制氢铁催化剂之一。
相对于镍和钴,简单高效的含铁制氢催化剂的报道并不多,有待于进一步研究开发。本文基于多吡啶配体L合成了一种FeIII配合物FeLCl3(1),考察了其在不同酸性强度的介质中(乙酸、三氟乙酸、四氟硼酸)电催化还原质子制氢的性能。
1 实验部分
1.1 仪器与试剂
试剂:三氟乙酸(GC级)、四氟硼酸-乙醚溶液(质量分数 55%)、冰乙酸(AR)、吡啶-2-甲醛(AR)、硼氢化钠、溴丙炔(AR)、叠氮甲基苯(AR)、六水合氯化铁(FeCl3·6H2O)以及其他试剂均购于国药集团。乙腈、四氢呋喃等(HPLC级)购于阿拉丁试剂公司,且使用前经过进一步纯化处理。
仪器:1H核磁共振谱数据是样品溶解在CDCl3经Varian 400记录。本文的电化学实验由Autolab 128N电化学工作站(瑞士万通)完成,实验在高纯干燥N2气氛条件下进行,采用经典的三电极体系,条件参数如下:参比电极为Ag/AgCl电极(0.45 mol/L[NBu4]BF4+0.05 mol/L[NBu4]Cl DCM 溶液),工作 电 极 玻 碳 电 极 (φ=1 mm),辅 助 电 极 铂 丝 (φ=1 mm),电解质TBABF40.1 mol/L MeCN溶液,扫描速率0.1 V/s,室温(25℃),N2气氛。待测样品物质的量在1~8 mmol,具体量根据实验要求,所用溶剂的体积为3.5~4.0 mL。如没有说明,所有电化学实验在室温下进行,电位都已经过二茂铁校正。
1.2 配合物1的合成与表征
配合物1的合成路线如图1所示。产物a、b和配体L的1H核磁共振数据如图2所示。
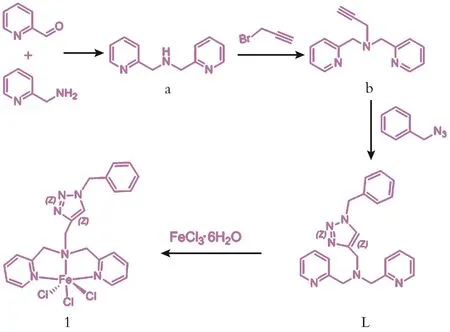
图1 配体L和配合物1的合成Fig.1 Synthesis of ligand L and complex 1
a的合成:取药品2-氨甲基吡啶(5.0 mL,0.052 mmol)与吡啶-2-甲醛(5.4 mL,0.052 mmol)在 500 mL的带支口的烧瓶中反应,以甲醇(AR)作为反应试剂,在室温环境搅拌8 h。然后再称取硼氢化钠(2.423 g,63.76 mmol),在低温冰盐浴中慢慢将其滴入反应瓶中,室温下反应12 h。当该反应结束,把溶剂抽干浓缩,加10 mL去离子水震荡,用二氯甲烷萃取,收集有机相。随后饱和食盐水震荡洗涤4次,无水硫酸镁干燥4 h。过滤、收集滤液浓缩至3~4 mL,过柱 (展开剂体积比V(EA)/V(EtOH)/V(Et3N)=5∶1∶0.1)分离得到产物 a(6.0 g,收率为 48%),核磁表 征 谱 图 如 图 2(a)所 示 。1H-NMR(400 MHz,CDCl3)δ 8.4(d,2H,Py),7.5(t,2H,Py),7.2(s,2H,Py),7.0(m,2H,Py),3.9(s,4H,CH2),2.8(s,1H,NH)。
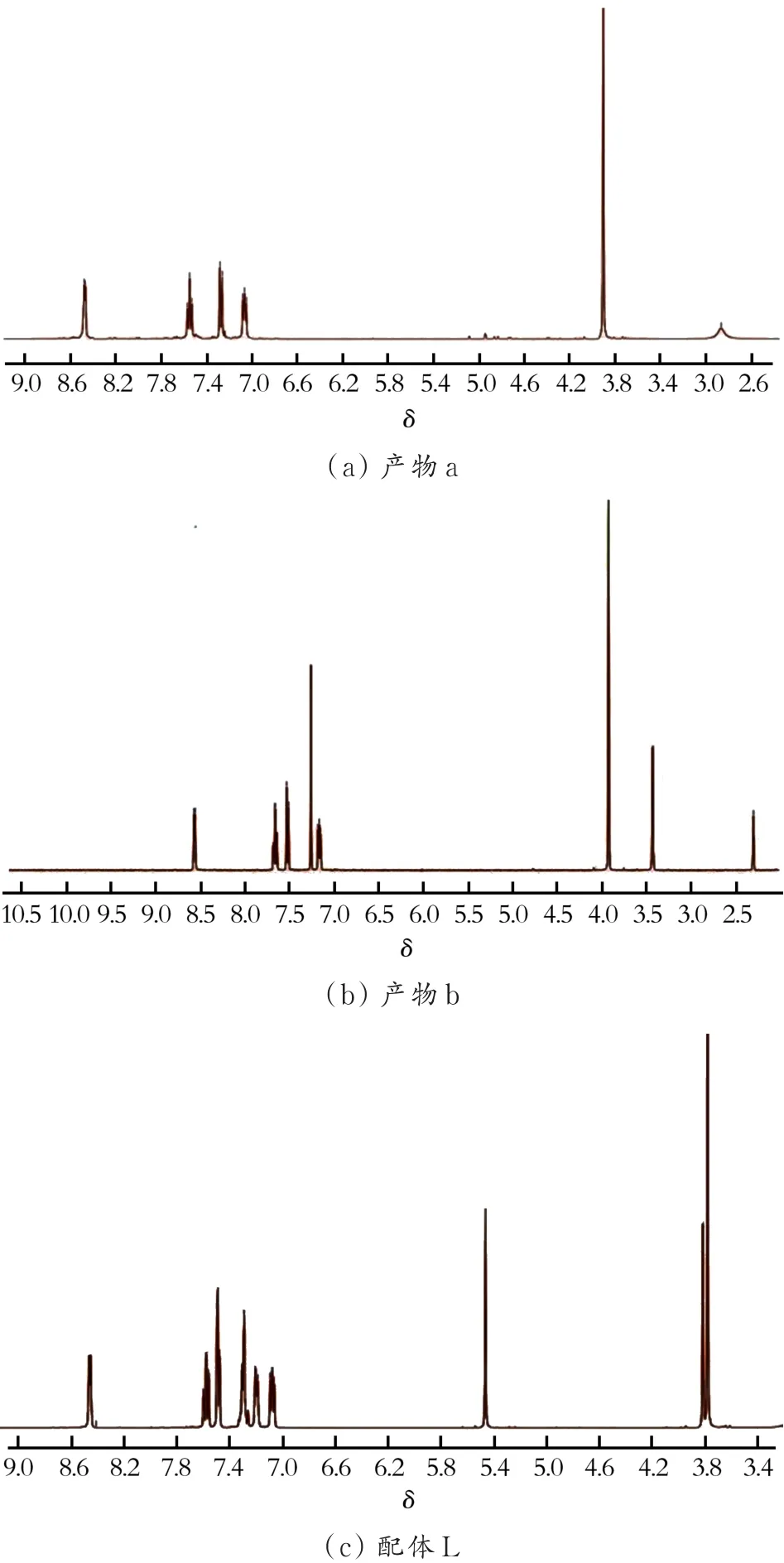
图2 产物a、b和配体L的1H核磁共振Fig.2 1H NMR data for a,b and ligand L
b 的 合 成 :上 步 产 物 a(0.95 g,5 mmol)溶 于THF,置于100 mL支口烧瓶中,称取过量(约4倍)的碳酸钾(2.76 g,20 mmol)加入烧瓶中,再取等物质的量的溴丙炔(0.4 mL,5 mmol)加入反应瓶中,室温条件搅拌12 h停止反应,过滤后的滤液浓缩至3~4 mL,过柱分离(展开剂体积比 V(EA)/V(EtOH)/V(Et3N)=15∶1∶0.1)得到黄色的似油状产物 b(0.9 g,收率为 80%),核磁表征谱图如图 2(b)所示 。1H-NMR(400 MHz,CDCl3)δ 8.5(d,2H),7.6(m,2H),7.5(d,2H),7.1(m,2H),3.90(s,5H),3.92(d,4H),3.4(s,2H),2.3(s,1H)。
配体L的合成:氩气气氛下,取上步产物b(293 mg,0.735 mmol)和 叠 氮 甲 基 苯 (246.5 mg,1.86 mmol),分别用无水无氧纯化处理后的四氢呋喃(HPLC)溶解,加入250 mL支口烧瓶中。随后称取干燥的CuI(35.5 mg,0.186 mmol)及除氧的三乙胺(0.25 mL,1.855 mmol)置于反应瓶中使其发生反应,CuI固体的逐渐溶解是判断该反应是否发生配位反应的关键。此反应是在45℃下搅拌20 h左右停止反应,过滤、浓缩,过柱分离(展开剂体积比V(EA)/V(EtOH)/V(Et3N)=15∶1∶0.1)得 到 油 状 物(237.5 mg,收率为 48%),核磁表征谱图如图 2(c)所示。1H-NMR(400 MHz,CDCl3)δ 8.4(d,2H,Py),7.55(tt,2H,Py),7.51~7.39(m,3H,Ph),7.33~7.21(m,2H,Py,1H,CH),7.22~ 7.13(m,2H,Py),7.06(dd,2H,Ph),5.45(s,2H,CH2), 3.79(d,2H,CH2),3.76(s,4H,CH2).
配合物1的合成:取等物质的量的配体L(370.2 mg,1.0 mmol)和 FeCl3·6H2O(270.1 mg,1.0 mmol)分别溶解在甲醇中,先将配体L的甲醇溶液置于反应瓶中,然后在磁力搅拌下将三价铁的甲醇溶液缓慢滴加以保证完全反应,在滴加过程中有固体沉淀产生,室温条件搅拌4 h,利用TLC检测配体L是否反应完全。反应结束后,真空旋蒸除溶剂甲醇(留存少量),过滤得到不纯固体配合物1(280 mg,收率为68%),用有机溶剂洗涤固体产物。通过乙腈-乙醚两相液液扩散的方法提纯配合物1。元素分析结果:配合物1(C22H22Cl3FeN6,相对分子质量为532.65);元素理论计算值(质量分数,下同):C 49.61%,H 4.16%,N 15.78%;实际测试值:C 49.82%,H 4.29%,N 15.85%.
2 结果与讨论
2.1 化合物1的电化学性质
通过循环伏安法研究了铁配合物的电化学行为。在乙腈溶液中(室温,扫描速率为0.1 V/s,配合物浓度为3.2 mmol/L)对Fe配合物的循环伏安谱曲线,如图3所示。
从图3中可知,它包含两个还原峰和两个氧化峰。其中,峰电位在-0.35 V处的还原峰归属于FeIIFeII/FeIFeI过程,是一个两电子可逆还原过程;其对应的氧化峰电位在-0.15 V左右,由于配合物1的次配位距离二价铁中心较远,对其影响较小。而在峰电位低于-2.0 V的还原峰归于FeIFeI/Fe0Fe0的过程。总之,次配位环境对于二价铁中心的电子云会产生一定的影响,其影响大小主要与供电子能力以及距离二价铁中心的远近有关。

图3 配合物1在0.1 mol/L的[NBu4]BF4-CH3CN中的循环伏安曲线Fig.3 Cyclic voltammograms of complex 1 in 0.1 mol/L[NBu4]BF4-CH3CN
2.2 配合物1在不同质子酸中的电化学性质
为了研究配合物的催化质子还原性能,分别研究了不同强度酸(乙酸HAc、三氟乙酸TFA、四氟硼酸HBF4)条件下的电化学情况(室温,扫描速率为0.1 V/s)。在逐渐加酸的当量过程中,记录了乙腈溶液中配合物1的循环伏安曲线(CV)。在析氢反应中催化活性通常以不可逆波的出现为判据,其随着酸浓度的增加而增长,而峰的位置可用于评估超电位。配合物1分别在醋酸、三氟乙酸和四氟硼酸介质中随不同当量酸浓度增加的循环伏安曲线如图4所示。
对比图4中的循环伏安曲线可知,加入酸性介质后配合物1的还原峰(FeIII/FeII)峰电位均正向移动,并且还原峰由原来的可逆过程变成不可逆过程,认为这种在酸存在下电位发生移动是由于质子耦合电子转移(PCET)。仔细观察可知,配合物1向阳极方向移动的距离较大,这很可能是由于吡啶上N原子的质子化降低了配合物1的还原峰峰电位引起的。
从配合物1的还原峰可以明显看出,配合物随着酸浓度的增加,还原峰的峰电流强度均增加。同时随着酸强度的增加,其在-1.4~-2.0 V出现了新的还原峰,且峰电流强度也随着酸浓度的增加不断增加,这是由于配合物1中存在可质子化的N原子使其具有电化学还原质子催化活性。而在加入质子酸后产生的新还原峰,是接受质子后的质子化产物获得一个电子形成中间产物,然后可继续从周围环境中得到质子,从而继续发生还原过程产生新的还原峰。
对比强酸和弱酸的循环伏安曲线结果,可得配合物的电化学还原质子催化活性,不仅与其自身性质有关,还与加入的质子酸的强弱有关。加入HAc(弱酸)时在-2.0 V处新形成较宽的还原峰,而加入强酸(TFA和HBF4)时,新出现两个还原峰(-1.8 V和-1.4 V),这与酸的供质子能力有关。

图4 配合物1在不同当量HAc、TFA和HBF4的循环伏安曲线Fig.4 Cyclic voltammograms of Complex 1 in different equivalent HAc,TFA and HBF4
观察新形成的还原峰,发现其还原峰峰电位都有随着质酸酸浓度的增加向正电位方向移动的趋势,并且峰电流均明显增加,说明质子浓度增加,配合物电催化还原活性增强。仔细比较图4(a)-(c),还可发现FeIII/FeII的还原峰峰电位与质子酸酸强度有关,随着酸强度的增加,其向正方向移动的幅度有所不同,四氟硼酸>三氟乙酸>乙酸,并且在四氟硼酸体系中还原峰(FeIII/FeII)峰电位由可逆过程变成不可逆过程。
2.3 催化机理的探讨
图5给出了铁配合物催化剂电催化析氢反应机理。

图5 可能的催化机理Fig.5 Proposed catalytic mechanism
由图5可见,反应关键步骤是铁配合物催化剂FeII电还原至FeI后,继续发生质子化生成FeIII-H中间体。析氢过程可以通过FeIII-H的质子化或者两个FeIII-H中间体进行双分子反应而发生。也可以将FeIII-H进一步还原成FeII-H,然后通过类似的均裂或者异裂反应产生氢气[17-18]。
Hammes-Schiffer和Muckerman的理论计算结果也表明[19-20],催化过程可能涉及到通过还原FeIIIH产生FeII-H中间体的机理。本文研究工作所得实验结果为电催化还原析氢反应过程中可形成FeII-H中间体提供了有力实验证据。此外,共价连接的吡啶N原子的配合物电化学研究表明,氢析出速率与单体类似物生成的速率有不可忽略的联系。
3 结 论
合成了多吡啶配体配位的铁配合物,并进行了一系列表征,成功制备了FeIII配合物。借助电化学循环伏安法进行其氧化还原性能的研究,进行了不同强度质子酸(乙酸、三氟乙酸、四氟硼酸)的电催化实验,研究了配合物的电催化质子还原性能。结果表明,在[NBu4]BF4-CH3CN溶液中,配合物对不同强度酸的质子催化有一个共同点,即在加入1当量质子源时,配合物的第一个还原峰电流强度均有增加。而后随质子源浓度增加,峰电流强度增加较为缓慢,直至后来影响趋于不变;-2.0~-1.4 V电位处的峰电流增加较为明显。当以强酸四氟硼酸作为质子源时,出现了两个新的还原峰,且新还原峰的峰电流随着酸当量浓度的增加而明显变化。说明以强酸作质子源时,配合物的电催化制氢效果更加明显。同时推测了该配合物可能的催化机理过程。
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
科学导报(2022年21期)2022-04-10
中学生数理化(高中版.高二数学)(2020年1期)2020-02-20
北京大学学报(自然科学版)(2019年6期)2019-11-27
分析化学(2018年12期)2018-01-22
成长·读写月刊(2017年3期)2017-04-08
中学生理科应试(2017年2期)2017-04-01
分析化学(2017年1期)2017-02-06
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
科技视界(2016年26期)2016-12-17
